De novo mutácie. Nové (De Novo) mutácie môžu byť detekované v oplodnených embryách pomocou preimplantačnej genetickej diagnostiky (PGD). Klinické charakteristiky Noonanovho syndrómu
Amniocentéza - test používaný na získanie vzorky na analýzu génov a chromozómov plodu. Plod je v maternici obklopený tekutinou. Táto tekutina obsahuje malý počet kožných buniek nenarodeného dieťaťa. Malé množstvo tekutiny sa odoberie tenkou ihlou cez brušnú stenu matky (brucho). Kvapalina sa posiela do laboratória na testovanie. Viac informácií nájdete v brožúre Amniocentéza.
Autozomálne dominantné genetické ochorenie je choroba, pri ktorej musí človek zdediť jednu zmenenú kópiu génu (mutáciu) od jedného zo svojich rodičov. Pri tomto type dedičnosti sa choroba prenáša na polovicu detí páru od jedného z rodičov, ktorý je chorý. Obe pohlavia sú rovnako pravdepodobne postihnuté. V rodinách existuje vertikálny prenos choroby: od jedného rodiča po polovicu detí.
Autozomálne recesívne genetickéchoroba - Ide o ochorenie, pri ktorom musí človek zdediť dve zmenené kópie génu (mutáciu), jednu od každého rodiča. Pri tomto type dedičstva je chorá štvrtina detí páru. Rodičia sú zdraví, no sú prenášačmi choroby. Osoba, ktorá má iba jednu kópiu zmeneného génu, bude zdravým nosičom. Ďalšie informácie nájdete v brožúre Recesívna dedičnosť.
Autozomálne - vlastnosť, ktorej gén sa nachádza na autozómoch.
Autozómy -Ľudia majú 23 párov chromozómov. Páry 1 až 22 sa nazývajú autozómy a vyzerajú rovnako u mužov a žien. Chromozómy 23. páru sa u mužov a žien líšia a nazývajú sa pohlavné chromozómy.
Biopsia choriových klkov, FVP - postup vykonávaný počas tehotenstva na odber buniek z plodu na testovanie génov alebo chromozómov nenarodeného dieťaťa na určité dedičné stavy. Malý počet buniek sa odoberie z vyvíjajúcej sa placenty a pošle sa do laboratória na testovanie. Viac informácií nájdete v brožúre Biopsia choriových klkov.
vagína - orgán spájajúci maternicu s vonkajším prostredím, pôrodné cesty.
gén - informácie potrebné pre fungovanie organizmu, uložené v chemická forma(DNA) na chromozómoch.
Genetické - spôsobené génmi, súvisiace s génmi.
Genetický výskum - test, ktorý môže pomôcť určiť, či existujú zmeny v jednotlivých génoch alebo chromozómoch. Viac informácií nájdete v brožúre Čo je genetické testovanie?
Genetické ochorenie - ochorenie spôsobené abnormalitami v génoch alebo chromozómoch.
vymazanie - strata časti genetického materiálu (DNA); tento termín sa môže použiť na označenie straty časti génu aj chromozómu. Viac informácií nájdete v brožúre Chromozomálne poruchy.
DNA - chemická látka, z ktorej sa skladajú gény a ktorá obsahuje informácie potrebné na fungovanie organizmu.
Duplikácia - abnormálne opakovanie sekvencie genetického materiálu (DNA) v géne alebo chromozóme. Viac informácií nájdete v brožúre Chromozomálne poruchy.
Meranie hrúbky golierového priestoru (TVP) - Ultrazvukové vyšetrenie zadnej časti krčnej oblasti plodu, ktorá je na začiatku tehotenstva naplnená tekutinou. Ak má dieťa vrodené ochorenie (napríklad Downov syndróm), hrúbka nuchálnej translucencie sa môže zmeniť.
Inverzia - zmena v sekvencii génov na jednom chromozóme. Viac informácií nájdete v brožúre Chromozomálne poruchy.
Vloženie - vloženie dodatočného genetického materiálu (DNA) do génu alebo chromozómu. Viac informácií nájdete v brožúre Chromozomálne poruchy.
karyotyp - popis chromozómovej štruktúry jednotlivca vrátane počtu chromozómov, počtu pohlavných chromozómov (XX alebo XY) a akýchkoľvek odchýlok od normálneho počtu.
Bunka-Ľudské telo pozostáva z miliónov buniek, ktoré slúžia" stavebné bloky" Bunky na rôznych miestach ľudského tela vyzerajú odlišne a vykonávajú rôzne funkcie. Každá bunka (okrem vajíčok u žien a spermií u mužov) obsahuje dve kópie každého génu.
Prstencový chromozóm je termín, ktorý sa používa, keď sa konce chromozómov spoja a vytvoria kruh. Ďalšie informácie nájdete v brožúre Chromozomálne translokácie.
maternica -časť ženského tela, v ktorej počas tehotenstva rastie plod.
Lekárske genetické poradenstvo- informácie a lekárska pomoc ľuďom, ktorých sa týka prítomnosť stavu v rodine, prípadne dedičného charakteru.
Mutácia- zmena v sekvencii DNA špecifického génu. Táto zmena v sekvencii génu vedie k tomu, že informácie v ňom obsiahnuté sú narušené a nemôže správne fungovať. To môže viesť k rozvoju genetického ochorenia.
Potrat - p Predčasné ukončenie tehotenstva, ku ktorému dochádza skôr, ako je dieťa schopné prežiť mimo maternice.
Nevyvážená translokácia - translokácia, pri ktorej chromozomálna prestavba vedie k získaniu alebo strate určitého množstva chromozomálneho materiálu (DNA), alebo súčasne k získaniu ďalšieho a strate časti pôvodného materiálu. Môže sa vyskytnúť u dieťaťa, ktorého rodič je nosičom vyváženej translokácie. Ďalšie informácie nájdete v brožúre Chromozomálne translokácie.
Nosič chromozomálnej prestavby - osoba, ktorá má vyváženú translokáciu, pri ktorej sa množstvo chromozomálneho materiálu ani neznižuje, ani nezvyšuje, čo zvyčajne nespôsobuje zdravotné problémy.
Prepravca - osoba, ktorá zvyčajne nemá ochorenie (momentálne), ale je nositeľom jednej zmenenej kópie génu. V prípade recesívneho ochorenia býva nosič zdravý; pri dominantnom ochorení s neskorým nástupom človek ochorie neskôr.
Hnojenie - splynutie vajíčka a spermie za vzniku prvej bunky dieťaťa.
Placenta- orgán priliehajúci k vnútornej stene maternice tehotnej ženy. Plod dostáva živiny cez placentu. Placenta vyrastá z oplodneného vajíčka, takže obsahuje rovnaké gény ako plod.
Pozitívny výsledok - výsledok testu, ktorý ukazuje, že testovaná osoba má zmenu (mutáciu) v géne.
Pohlavné chromozómy - chromozóm X a chromozóm Y. Súbor pohlavných chromozómov určuje, či je jedinec muž alebo žena. Ženy majú dva chromozómy X, muži jeden chromozóm X a jeden chromozóm Y.
Prediktívne testovanie - genetické testovanie na identifikáciu stavu, ktorý sa môže alebo rozvinie počas života. Keď je genetický test zameraný na identifikáciu stavu, ktorý sa v budúcnosti takmer určite vyvinie, nazýva sa test presymptomatické.
Prenatálna diagnostika- štúdia vykonaná počas tehotenstva na zistenie prítomnosti alebo neprítomnosti genetického ochorenia u dieťaťa.
Recipročná translokácia - translokácia, ku ktorej dochádza, keď sa dva fragmenty odlomia z dvoch rôznych chromozómov a zmenia miesto. Ďalšie informácie nájdete v brožúre Chromozomálne translokácie.
Robertsonova translokácia - nastáva, keď sa jeden chromozóm pripojí k druhému. Ďalšie informácie nájdete v brožúre Chromozomálne translokácie.
Vyvážená translokácia – t relokácia (chromozomálna prestavba), pri ktorej sa množstvo chromozomálneho materiálu neznižuje ani nezväčšuje, ale presúva sa z jedného chromozómu na druhý. Človek s vyváženou translokáciou tým zvyčajne netrpí, ale riziko vzniku genetických chorôb u jeho detí je zvýšené. Ďalšie informácie nájdete v brožúre Chromozomálne translokácie.
Stav prilepený k podlahe- Pozri dedičnosť viazanú na X.
Spermie - otcovská zárodočná bunka, otcovský príspevok k vytvoreniu bunky, z ktorej sa vyvinie bábätko. Každá spermia obsahuje 23 chromozómov, jeden z každého páru chromozómov otca. Spermia sa spojí s vajíčkom a vytvorí prvú bunku, z ktorej sa vyvinie nenarodené dieťa.
Premiestnenie - preskupenie chromozomálneho materiálu. Vyskytuje sa, keď sa fragment jedného chromozómu odlomí a pripojí sa na iné miesto. Ďalšie informácie nájdete v brožúre Chromozomálne translokácie.
Ultrazvukové vyšetrenie (ultrazvuk) - bezbolestný test, ktorý pomocou zvukových vĺn vytvorí obraz o raste plodu v maternici matky. Dá sa to urobiť pohybom hlavy skenera pozdĺž povrchu brušnej steny matky (žalúdka) alebo vo vnútri vagíny.
Chromozómy - vláknité štruktúry viditeľné pod mikroskopom, ktoré obsahujú gény. Normálne má človek 46 chromozómov. Jednu sadu 23 chromozómov zdedíme od našej matky a druhú sadu 23 chromozómov od nášho otca.
X-viazané ochorenie- genetické ochorenie, ktoré vzniká v dôsledku mutácie (zmeny) génu umiestneného na X chromozóme. Ochorenia viazané na X zahŕňajú hemofíliu, Duchennovu svalovú dystrofiu, syndróm fragilného X a mnohé ďalšie. Ďalšie informácie nájdete v brožúre Dedičstvo viazané na X.
XX- takto je zvyčajne reprezentovaný ženský súbor pohlavných chromozómov. Normálne má žena dva chromozómy X. Každý chromozóm X je zdedený od jedného z rodičov.
X chromozóm - Jeden z pohlavných chromozómov. Ženy majú zvyčajne dva X chromozómy. Muži majú zvyčajne jeden chromozóm X a jeden chromozóm Y.
Vaječník/vaječníky- orgány v tele ženy produkujúce vajíčka.
vajíčko - reprodukčná bunka matky, ktorá bude slúžiť ako základ pre vytvorenie prvej bunky nenarodeného dieťaťa. Vajíčko obsahuje 23 chromozómov; jeden z každého páru, ktorý má matka. Vajíčko sa spojí so spermiou a vytvorí prvú bunku dieťaťa.
De novo - od latinský výraz znamenajúci „znovu“. Používa sa na popis zmien v génoch alebo chromozómoch (mutáciách), ktoré sa novotvoria, t.j. ani jeden z rodičov osoby s mutáciou de novo nemá tieto zmeny.
XY- takto je zvyčajne reprezentovaná mužská sada pohlavných chromozómov. V Norii majú muži jeden chromozóm X a jeden chromozóm Y. Muži dedia chromozóm X od svojej matky a chromozóm Y od svojho otca.
Y chromozóm- jeden z pohlavných chromozómov. Normálne majú muži jeden chromozóm Y a jeden chromozóm X. Žena má zvyčajne dva chromozómy X.
Z BrainstormWiki
Čo je mutácia?
Začnime tým, že neexistuje žiadny „správny“ génový kód. Preto, ak ja mám jeden gén a ty iný, neznamená to, že jeden z nás je mutant.
Existujú však zmeny v genetickom kóde, ktoré vedú k zjavným problémom, a tie sú zvyčajne zriedkavé; nazývajú sa mutácie. Zmeny, ktoré sa vyskytujú u viac ako 1% populácie, sa správnejšie nazývajú nie mutácie, ale polymorfizmy.
Mutácie môžu byť dedené od rodičov, alebo sa môžu vyskytnúť iba u dieťaťa - v takom prípade sa nazývajú de novo
Mutácie môžu nastať pri vývoji organizmu, pri delení a diferenciácii buniek – ide o tzv somatické mutácie; nemožno ich zdediť, pretože „žijú“ v bunkách tela (soma), ale nie v zárodočných bunkách.
Naopak, mutácie, ktoré ovplyvňujú zárodočné bunky, sa dedia a sú tzv zárodočné mutácie
Mutácie môžu byť tichý- sú tam, ale nemajú žiadny účinok. Ide o drvivú väčšinu – ak si pamätáte, gény zaberajú len menej ako 2 % všetkej DNA. Každý z nás je nositeľom minimálne desiatok takýchto mutácií. Ak padnú na nedôležité úseky DNA, nikto si nič nevšimne.
Mutácie môžu ovplyvniť jedno „písmeno“ génového kódu - alebo môžu ovplyvniť celý obrovský fragment DNA. Tento fragment môže byť najčastejšie stratený (delecia) alebo duplikovaný (duplikácia) - v tomto prípade bunky zmenia množstvo proteínov syntetizovaných pre postihnuté gény - zmení sa ich dávka
Pre rôzne typy mutácií existujú odlišné typy analýzy a testy a my sa ich teraz pokúsime prejsť.
Od veľkých po malé
Aneuploidia - abnormalita v počte chromozómov
Najväčšou genetickou anomáliou je zmena počtu chromozómov. Hoci toto nie je autizmus, oplatí sa začať s. Pred 50 rokmi sa zistilo, že Downov syndróm spôsobujú tri 21 chromozómy namiesto dvoch. Toto sa nazýva trizómia; a zmena vo všeobecnom počte chromozómov je aneuploidia. Okrem toho existuje trizómia 13 chromozómov (Patauov syndróm) a 18 (Edwardsov syndróm), ako aj množstvo aneuploidií pohlavných chromozómov (X a Y)
To všetko je možné vidieť bežným optickým mikroskopom - analýzou nazývanou „karyotypizácia“. Chromozómy deliacich sa buniek sú fotografované, triedené a opísané. Na obrázku je karyotyp ženy s tromi 21 chromozómami.
Účinok trizómie je obrovský a viditeľný v mnohých procesoch: v štruktúre tela, vo fyziologických procesoch a dokonca aj v charakteristických proteínoch v krvi nosiacej matky (takto teraz fungujú skríningové testy v dedine Dauna)
Prečo sa nachádzajú iba trizómie 21, 13 a 18 a pohlavné chromozómy? To sa môže stať s ktorýmkoľvek chromozómom, ale zvyšok neprežije ani do štádia nápadného tehotenstva. Možno dôvodom je, že chromozómy 21, 13 a 18 patria medzi najchudobnejšie na proteínové gény (nezabudnite, sú očíslované podľa veľkosti, sú aj malé) a zvyšovanie ich dávky je relatívne tolerovateľné. Potvrdzuje to nasledujúca tabuľka: v počiatočných fázach vývoja sú možné akékoľvek aneuploidie, a keď sa blížite k úspešnému pôrodu, iba tieto 3.

Kopírovať variáciu čísla

CNV je tiež buď duplikácia alebo absencia určitej časti DNA, ale nie na úrovni celého chromozómu, ale niekoľkých miliónov nukleotidov.
Na ich detekciu sa používa testovanie nazývané CGH array: komparatívna genómová hybridizácia, tiež FISH a iné.V našej praxi sa často nazývajú aj „molekulárna karyotypizácia“, microarray chromozomálna analýza atď.
Stále ide o obrovské kusy DNA, ktoré môžu obsahovať desiatky génov a riadiacich štruktúr. Účinky sú veľmi nápadné a takmer vždy zahŕňajú dysmorfizmy a zmeny kognitívnych schopností. Sú označené postihnutou oblasťou génového kódu, napríklad delécia 2q32 - strata úseku v pruhu 32 na dlhé rameno 2 chromozómy (zapamätajte si časť 1 a „adresy“ chromozómových úsekov) Sem patrí aj mnoho známych „named“ syndrómov, napríklad Williamsov syndróm – delécia 7q11.23
Predpokladá sa, že takéto mutácie CNV vysvetľujú 3 % až 10 % prípadov autizmu. Toto je prakticky jediný typ genetickej abnormality, ktorá je s istotou spojená s niektorými - syndrómovými - formami autizmu.
Ešte raz, hlavná vec, ktorú potrebujete vedieť o CNV pri autizme: fungujú v malej časti prípadov, ale ich účinok je viditeľný v mnohých aspektoch, od dysmorfizmov po inteligenciu. Tie. Prítomnosť defektov v CNV možno takmer vždy vidieť buď priamo na tvári, alebo vo forme celkom zjavných porúch, ako je systémová hypotenzia a ataxia...
Niektoré známe a zdokumentované CNV

Prítomnosť údajov CNV však nezaručuje autizmus ako diagnózu. Prienik nikdy nie je 100%. Nedávna estónska štúdia na skupine ľudí bez akejkoľvek diagnózy ukázala, že nosiči CNV 16p11.2 vykazovali dysmorfizmy hlavy, obezitu, kognitívne poruchy – ale nemali diagnózu autizmu, na rozdiel od údajov v tabuľke vyššie;
Existuje online databáza CNV, ktoré spôsobujú autizmus a súvisiace syndrómy http://projects.tcag.ca/autism/
Syndromový autizmus
Syndromový autizmus sa nazýva, keď sa verí, že autistické symptómy sú spôsobené pochopiteľnou genetickou poruchou - zvyčajne CNV, ale nielen (pozri napríklad Fragile X). Mnohí vedci tvrdia, že tento termín je nesprávny. Pravdepodobne by bolo správnejšie povedať „autizmus pochopiteľnej genetickej etiológie“ alebo niečo podobné.
Veľmi dobrá recenzia najznámejšie formy autizmu s „pochopiteľnou genetickou etiológiou“ – t.j. Syndromové formy autizmu nájdete na blogu Emily Casanovovej
- Časť 1: Timothyho syndrómy, Smith-Lemli-Opitz, CHARGE, Cornelia de Lange, Lujan-Fryns
Ešte raz o penetrácii
Penetrácia je termín, ktorý označuje percento nosičov mutácie, ktorá prejavuje (v tomto prípade) autizmus. Prehľad v predchádzajúcej kapitole ukazuje, že penetrácia syndrómových foriem autizmu je 60 až 90 percent.
92% penetrancia pre idic(15) je opísaná ako "ohromujúca". Áno, to je veľa. To je viac než dosť na to, aby sa tento genetický variant považoval za príčinu autizmu – ak sa zistí u dieťaťa a príznaky sú konzistentné. To tiež dáva dôvod domnievať sa, že ak je táto mutácia vylúčená v PGD, ďalšie dieťa tí istí autisti rodičia nebudú existovať.
SNP

Teraz poďme k najmenším genetickým variáciám obsiahnutým len v jednom nukleotide, jednom „písmene.“ Existuje niekoľko miliónov bodov v ľudskom genóme, kde sú variácie v jednom „písmene“ génového kódu bežné; Všetko naokolo je stabilné, no na týchto špecifických miestach je to pre rôznych ľudí iné. Navyše, z nejakého dôvodu sú takéto variácie takmer vždy bialelické, to znamená, že existujú len dva varianty „písmen“ zo štyroch a sú prítomné vo veľmi veľkej časti populácie – jeden variant je často v 60 % a druhý v 40 %. Toto nie je mutácia, ale polymorfizmus, ak si pamätáte časť 7. (aby bolo SNP oficiálne uznané, jeho prevalencia musí byť aspoň 1%)
To znamená, že nejde o „mutácie“, nie o chyby DNA. Toto je norma a jej variácie.
Takéto variácie sa nazývajú SNP (čítaj „ústrih“), označené číslami ako rs2320030, a bolo ich nájdených a popísaných asi 10 miliónov.
Štúdie asociácií celého genómu
Analýza, ktorá ukazuje, ktoré varianty SNP (alely) sú prítomné u konkrétnej osoby, sa vykonáva na čipe SNP. Je to platňa, na ktorú sú okolo populárnych SNP „vytlačené“ vzorky ľudského genómu, na túto platňu sa nanesie DNA testovaného subjektu (často vzorka slín) a potom sa pozrie, kde sa „prilepí“ a kde sa nedrží. 't. Populárna analýza 23andme robí práve to a poskytuje približne 1 milión takýchto SNP. Toto sa nazýva „genotypizácia“ a je pomerne lacné, preto sa v súčasnosti často (a často nevhodne) používa. Lacné, pretože SNP sú dobre známe a túto analýzu je vhodné robiť online vo veľkých množstvách.
Táto dostupnosť podnietila vznik špeciálneho typu výskumu, Genome-Wide Association Studies (GWAS), v rámci ktorého sa vyberie skupina nositeľov dedičného znaku a kontrolná skupina, všetci sú genotypovaní a potom sa zisťuje, či existuje je akékoľvek SNP, ktoré prvé majú v jednom variante a v druhom - iným spôsobom.
Problém je v štatistike: keďže existujú milióny SNP, je vysoká pravdepodobnosť, že uvidíme spojenie tam, kde žiadne nie je: len napríklad v skupine 30 autistov sa objaví SNP “ správne“ 30-krát. Ak hodíte mincou miliónkrát, pristane na hrane :) Tento problém sa dá vyriešiť pomocou štatistických metód a úspešne sa s ním vysporiadať. Vo všeobecnosti sa však výsledky GWAS pre autizmus nedostatočne replikujú.
Čo znamená prítomnosť SNP?

Vplyv jedného alebo druhého variantu SNP je zvyčajne zanedbateľný. Nemôže to byť inak, keďže všetky sú podľa definície v populácii veľmi bežné. Sú však ochorenia (napríklad cystická fibróza), pre ktoré sú rozhodujúce (zvyčajne však nie pri jednom SNP: zvyčajne prítomnosť „zlého“ variantu štatisticky zvyšuje riziko poruchy povedzme o 2 %).
Nezistilo sa ani jedno SNP, ktoré by sa nejakým významným spôsobom spájalo s autizmom. Mimochodom, tie, ktoré sú aspoň trochu spojené, nie sú spravidla v génoch, ale v riadiacich prvkoch DNA - t.j. nie v exome.
Preto všetky testy, ktoré používajú genotypizáciu SNP a vypočítavajú významné „riziko autizmu“, sú s najväčšou pravdepodobnosťou šarlatánstvo. Existuje pomerne málo stránok, ktoré ponúkajú stiahnutie výsledkov 23andme a získanie „personalizovaných“ receptov na liečbu autizmu. Najznámejší je „Dr.“ Yasko, ktorý propaguje recepty na „mutácie MTHFR“. šarlatánstvo. Ešte raz, toto nie sú mutácie. MTHFR je dôležitý gén v metabolizme folátu a pravdepodobne je spojený s autizmom, ale nie je na úrovni SNP, ktorý treba hľadať.
Aby sme boli spravodliví, treba povedať, že kombinácia SNP (stovky rôznych SNP, z ktorých každý má malý príspevok, ale pridáva niečo významné) sa zdá byť schopná vysvetliť schizofréniu (na rozdiel od autizmu) - čo je to, čo takmer tí istí ľudia sa snažia identifikovať projekt ľudského genómu a takmer rovnako herkulovské úsilie. Tam však hovoríme o stovkách útržkov.
De novo mutácie

„Skutočné“ mutácie, ktoré nežijú trvalo v populácii, mutácie, ktoré sa objavujú u konkrétnych jedincov a prenášajú sa na ich deti (alebo sa vyskytujú u detí), sa nazývajú mutácie de novo. Môžu to byť veľké oblasti (CNV), malé vloženia a vymazania (indely) alebo jednotlivé „písmená“. Tento príspevok je o posledných dvoch typoch.
Na ich odhalenie sú nevhodné testy na lacných čipoch, genóm treba postupne čítať, alebo, ako sa tomu hovorí, sekvenovať. Môžete si prečítať celý genóm – sekvenovanie celého genómu (drahé), aj len exóm, iba časť kódujúcu proteíny – sekvenovanie exómu (relatívne lacné, 1000 USD na osobu). V klinickej praxi sa teda takmer nikdy nepoužíva.
V zásade je takýchto de novo mutácií pomerne veľa – každý rodič ich dieťaťu odovzdá minimálne 100, no len malá časť pripadá na významné miesta v DNA. Nedávne, rešpektované publikácie (Iossifov a kol. 2012) odhadujú kľúčový príspevok mutácií de novo v približne 10 % prípadov autizmu. Ak ste boli na konferencii v máji, Nagwa Meguid hovorila o „Novom géne a jedinečnej forme autizmu“ – presne o takomto prípade, de novo mutácie (rôzne) v géne BCKDK.
Avšak aj v tomto jednoduchom príbehu: tu je mutácia, tu ovplyvňuje gén dôležitý pre funkciu mozgu, tu sme demonštrovali, že tento gén ovplyvňuje autizmus – je tu veľká záhada. Ak na rozdiel od Ivana Iosifova študujeme rodiny nie s jedným, ale s dvoma autistami, ako to tento rok urobili Yuen a kolegovia, potom sa ukáže, že sa im darí odoberať rôzne mutácie od tých istých rodičov.
Zhrnieme a klasifikujeme typy „mutácií“
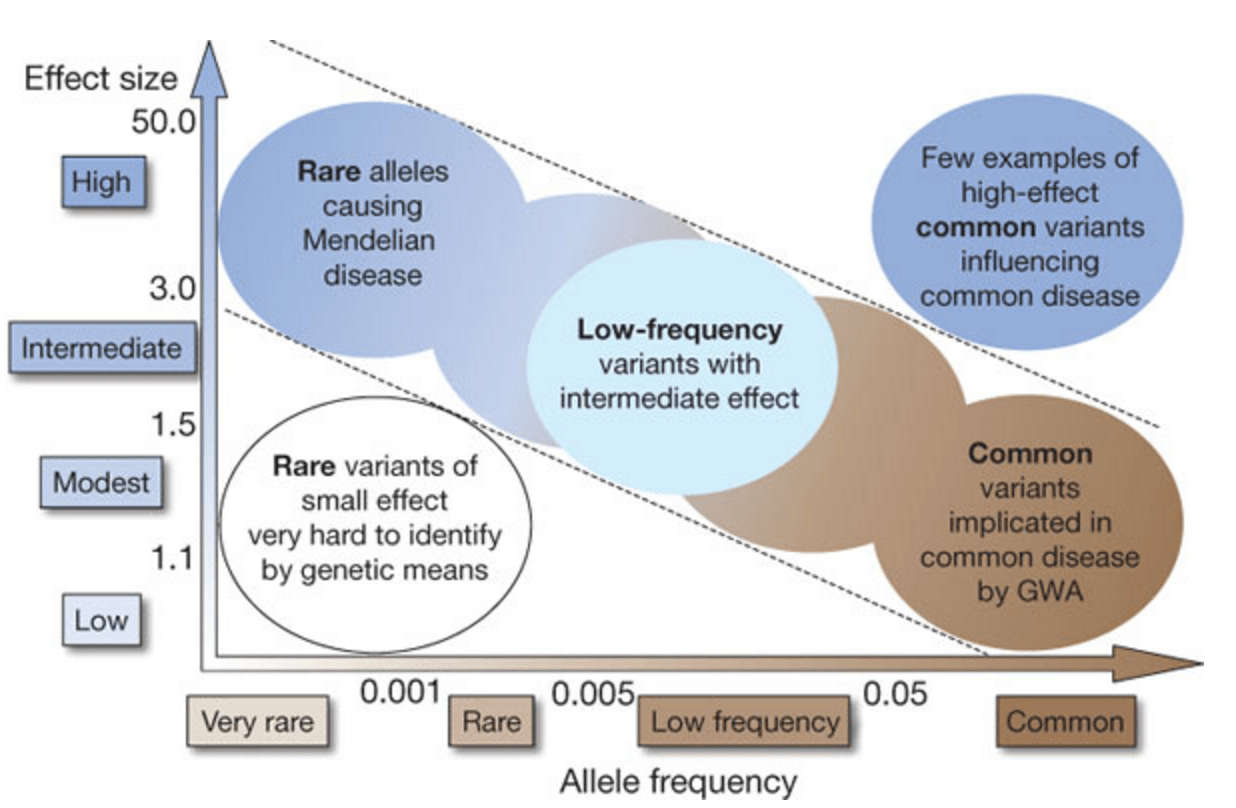
V predchádzajúcich častiach sme si to v krátkosti prešli rôzne druhy genetické varianty v DNA. Od veľkých rozmerov, zvyčajne s výrazným účinkom, až po veľmi malé.
A tu platí pravidlo: veľké anomálie sú zvyčajne zriedkavé, ale majú výrazný vplyv na mnohé aspekty fenotypu; malé varianty sú v populácii často bežné, ale každá z nich má zanedbateľný vplyv
Dá sa to zhrnúť do diagramu, ako je tento, kde horizontálna čiara predstavuje prevalenciu alely (genetický variant), od zriedkavej po bežnú, a vertikálna čiara je prejavený účinok u nosičov.
Je potrebné poznamenať, že pri autizme a schizofrénii ani tie „najsilnejšie“ CNV nemajú menedelovský efekt, čo zvyčajne zvyšuje riziko vzniku klinicky zistiteľného ochorenia z 1 % na 3 % – 10 %. To sa nazýva neúplná penetrácia. Môžete byť nositeľom známej „schizofrenickej CNV“, môžete mať dysmorfizmy a abnormality v endokrinnom systéme, ktoré sú pre ňu charakteristické, ale nemusíte mať schizofréniu, hoci môžete vykazovať určité subklinické abnormality.
V súlade s tým je individuálny účinok polymorfizmov typu SNP vo všeobecnosti zanedbateľný a nemá takmer žiadny praktický význam pre diagnostiku autizmu.
| Vzdelávací program o genetike autizmu |
|---|
| I. Vzdelávací program o genetike autizmu II. Genetika a dvojčatá III. Typy mutácií IV. |
Detekcia denovo mutácie v géne dystrofínu a jej význam pre medicínske genetické poradenstvo pri Duchennovej svalovej dystrofii
(klinické pozorovanie)
Muravleva E.A., Starodubova A.V., Pyshkina N.P., Duysenova O.S.
Vedecký školiteľ: doktor lekárskych vied Doc. Kolokolov O.V.
Štátna lekárska univerzita GBOU VPO Saratov pomenovaná po. IN AND. Razumovského ministerstvo zdravotníctva Ruskej federácie
Neurologická klinika Pedagogickej fakulty a PPS pomenovaná po. K.N. Treťjakov
Úvod. Duchennova svalová dystrofia (DMD) je jednou z najčastejších dedičných neuromuskulárnych chorôb. Jeho prevalencia je 2-5: 100 000 obyvateľov, populačná frekvencia je 1: 3500 novonarodených chlapcov. Túto formu svalovej dystrofie prvýkrát opísali Edward Meryon (1852) a Guillaume Duchenne (1861).
Ochorenie je charakterizované X-viazaným recesívnym typom dedičnosti a ťažkým, progresívnym priebehom. DMD je spôsobená mutáciou v géne pre dystrofín, ktorého lokus je lokalizovaný na Xp21.2. Asi 30% prípadov je spôsobených de novo mutáciami, 70% - nosičstvom mutácie matkou probanda. Dystrofín je zodpovedný za spojenie cytoskeletu každého svalového vlákna so základnou bazálnou laminou (extracelulárna matrica) prostredníctvom proteínového komplexu, ktorý pozostáva z mnohých podjednotiek. Absencia dystrofínu vedie k prenikaniu nadbytočného vápnika do sarkolemy ( bunková membrána). Svalové vlákna podliehajú nekróze, svalové tkanivo je nahradené tukovým tkanivom, ako aj spojivovým tkanivom.
Moderná diagnostika DMD je založená na posúdení súladu prejavov ochorenia s klinickými, anamnestickými a laboratórno-inštrumentálnymi (sérová kreatínkináza (SKK), elektroneuromyografia (ENMG), histochemické vyšetrenie svalovej biopsie) kritériami, genealogickou analýzou a molekulárne genetickými výskumné údaje.
Vykonávanie lekárskeho genetického poradenstva v mnohých rodinách dnes pomáha predchádzať narodeniu chorého dieťaťa. Prenatálna DNA diagnostika pre skoré štádia tehotenstva v rodinách s dieťaťom trpiacim DMD, umožní zvoliť rodičom ďalšiu taktiku a prípadne predčasné ukončenie tehotenstva, ak je ochorenie prítomné u plodu.
V niektorých prípadoch je klinický obraz pozorovaný u žien, ktoré sú heterozygotnými nosičmi mutantného génu vo forme zväčšených lýtkových svalov, miernej svalovej slabosti, znížených šľachových a periostálnych reflexov, podľa paraklinických štúdií sa zvyšuje hladina CCS. Okrem toho sa u žien so syndrómom Shereshevsky-Turner (genotyp 45, XO) môžu vyskytnúť klasické klinické prejavy DMD.
Klinický príklad. V našej ambulancii sa nachádza 7-ročný chlapec K., ktorý sa sťažuje na slabosť svalov rúk a nôh, únavu pri dlhšej chôdzi. Matka dieťaťa zaznamenáva pravidelné pády, ťažkosti pri chôdzi po schodoch, zhoršenú chôdzu (ako kačica), ťažkosti pri vstávaní zo sedu a zväčšenie objemu lýtkových svalov.
Raný vývoj dieťaťa prebiehal bez akýchkoľvek špeciálnych znakov. Vo veku 3 rokov jeho okolie zaznamenalo poruchy motorických funkcií vo forme ťažkostí pri chôdzi po schodoch, pri vstávaní, dieťa sa nezúčastňovalo vonkajších hier a začalo sa rýchlo unavovať. Potom sa chôdza typu „kačice“ zmenila. Ťažkosti sa zvýšili pri vstávaní zo sedu alebo z ľahu: krok za krokom „rebrík“ stúpajúci aktívnym používaním rúk. Postupne sa prejavil nárast objemu lýtka a niektorých ďalších svalov.
Pri neurologickom vyšetrení je vedúcim klinickým príznakom symetrická proximálna periférna tetraparéza, výraznejšia na nohách (svalová sila v proximálnych častiach horných končatín - 3-4 body, v distálnych častiach - 4 body, v proximálnych častiach dolné končatiny - 2-3 body, v distálnych častiach - 4 body). Chôdza sa zmenila na typ „kačacieho“. Používa pomocné („myopatické“) techniky, napríklad vstávanie s rebríkom. Svalový tonus je znížený, nie sú žiadne kontraktúry. Hypotrofia svalov panvového a ramenného pletenca. „Myopatické“ znaky, napríklad vo forme širokého medzilopatkového priestoru. Existuje pseudohypertrofia lýtkových svalov. Šľachové a periostálne reflexy - bez výrazného rozdielu medzi stranami; bicipitálny - nízky, tricipitálny a karporadálny - stredná živosť, koleno a Achillova - nízka. Na základe klinických údajov bolo podozrenie na DMD.
Pri štúdiu CCS bola jeho hladina 5379 jednotiek/l, čo je 31-krát viac ako norma (norma je až 171 jednotiek/l). Podľa ENMG boli zaznamenané znaky, ktoré sú charakteristické skôr pre stredne prebiehajúci primárny svalový proces. Získané údaje teda potvrdili prítomnosť DMD u pacienta.
Okrem probanda boli na vyšetrení aj jeho rodičia a staršia sestra. Žiadny z príbuzných probanda nemal klinické prejavy DMD. Matka však zaznamenala mierny nárast objemu lýtkových svalov. Podľa genealogického rozboru je proband jedinou chorou osobou v rodine. Nedá sa vylúčiť, že matka dieťaťa a sestra probanda sú heterozygotnými nosičmi mutantného génu (obr. 1).
Ryža. 1 Rodokmeň
V rámci lekárskeho genetického poradenstva bola v rodine K. vyšetrená prítomnosť/neprítomnosť delécií a duplikácií v géne pre dystrofín. Molekulárno-genetická analýza v laboratóriu DNA diagnostiky Moskovského štátneho výskumného centra Ruskej akadémie lekárskych vied odhalila deléciu exónu 45 u probanda K., čo napokon potvrdzuje stanovenú klinickú diagnózu DMD. Matka nemala u svojho syna identifikovanú deléciu exónu 45. Výsledkom analýzy bolo, že sestrina delécia exónu 45, identifikovaná u jej brata, sa nenašla. Preto u skúmaného človeka má mutácia s najväčšou pravdepodobnosťou de novo pôvod, ale môže byť aj výsledkom zárodočnej mozaiky u matky. V súlade s tým pri mutácii de novo bude riziko, že matka bude mať choré dieťa, určené populačnou frekvenciou tejto mutácie (1:3500, <1 %), čo je výrazne menej ako u recesívneho typu viazaného na X. dedičnosti (50 % chlapcov). Keďže nie je možné úplne vylúčiť, že mutácia môže byť výsledkom germinálnej mozaiky, pri ktorej dochádza k porušeniu dedičnosti podľa mendelovských zákonov, odporúča sa prenatálna diagnostika v ďalšom tehotenstve u matky a sestry probanda.
Záver. V súčasnosti má lekár široký arzenál symptomatických liekov používaných v liečbe DMD, napriek pokroku vedy však etiologická liečba DMD ešte nebola vyvinutá, účinné lieky Náhradná liečba DMD neexistuje. Podľa nedávneho výskumu kmeňových buniek existujú sľubné vektory, ktoré dokážu nahradiť poškodené svalové tkanivo. V súčasnosti je však možná len symptomatická liečba zameraná na zlepšenie kvality života pacienta. V tomto smere skorá diagnóza DMD hrá dôležitú úlohu pri včasnom medicínskom genetickom poradenstve a výbere ďalšej taktiky plánovaného rodičovstva. Na prenatálnu diagnostiku DNA sa odber choriových klkov (CVS) môže vykonať v 11. – 14. týždni tehotenstva, amniocentéza sa môže použiť po 15. týždni a odber krvi plodu sa môže vykonať približne v 18. týždni. Ak sa testovanie vykonáva na začiatku tehotenstva, predčasné ukončenie tehotenstva je možné, ak má plod ochorenie. V niektorých prípadoch sa odporúča vykonať predimplantačnú diagnostiku DNA a následne oplodnenie in vitro.
Závery. Na zabezpečenie včasnej detekcie a prevencie DMD je potrebné širšie využívať molekulárne genetické diagnostické metódy; zvýšiť pozornosť praktických lekárov v súvislosti s touto patológiou. Pri mutácii de novo je riziko matky mať choré dieťa určené populačnou frekvenciou mutácie génu dystrofínu. V prípadoch, keď je nosičom mutácie matka probanda, je na účely plánovania rodiny potrebná prenatálna alebo perimplantačná DNA diagnostika.
Lekárska cytogenetika je štúdium ľudského karyotypu za normálnych a patologických stavov. Tento smer vznikol v roku 1956, keď Tio a Levan zdokonalili metódu prípravy metafázových chromozómov a po prvýkrát stanovili modálny počet chromozómov (2n=46) v diploidnom súbore. V roku 1959 bola dešifrovaná chromozomálna etiológia mnohých chorôb - Downov syndróm, Klinefelterov syndróm, Shereshevsky-Turnerov syndróm a niektoré ďalšie syndrómy autozomálnej trizómie. K ďalšiemu rozvoju lekárskej cytogenetiky koncom 60. rokov 20. storočia prispel nástup metód diferenciálneho farbenia metafázových chromozómov, ktoré umožnili identifikovať chromozómy a ich jednotlivé oblasti. Diferenciálne metódy farbenia nie vždy zabezpečili správnu identifikáciu bodov zlomu v dôsledku štrukturálnych preskupení chromozómov. V roku 1976 Younis vyvinul nové metódy na ich štúdium v štádiu prometafázy, ktoré sa nazývali „metódy s vysokým rozlíšením“.
Použitie takýchto metód umožnilo získať chromozómy s rôznym počtom segmentov (od 550 do 850) a umožnilo identifikovať poruchy zahŕňajúce ich malé časti (mikroprestavby). Od začiatku 80. rokov 20. storočia. vstúpila ľudská cytogenetika nová etapa vývoj: chromozomálna analýza molekulárnych cytogenetických metód, do praxe zavedená fluorescenčná in situ hybridizácia (FISH - Fluorescence In Situ Hybridization). Táto metóda sa široko používa na detekciu jemnejších štrukturálnych abnormalít chromozómov, ktoré sa nedajú rozlíšiť diferenciálnym farbením. V súčasnosti používanie rôznych metód chromozomálnej analýzy umožňuje úspešne vykonávať pre- a postnatálnu diagnostiku chromozomálnych ochorení.
Chromozomálne ochorenia predstavujú veľkú skupinu klinicky rôznorodých stavov charakterizovaných mnohopočetnými vrodenými malformáciami, ktorých etiológia je spojená s kvantitatívnymi alebo štrukturálnymi zmenami v karyotype.
V súčasnosti sa rozlišuje takmer 1000 chromozomálnych abnormalít, z ktorých viac ako 100 foriem má klinicky definovaný obraz a nazývajú sa syndrómy; ich podiel na spontánnych potratoch, novorodeneckej úmrtnosti a chorobnosti je významný. Prevalencia chromozomálnych abnormalít medzi spontánnymi potratmi je v priemere 50 %, medzi novorodencami s ťažkými mnohopočetnými vrodenými chybami - 33 %, mŕtvo narodenými a perinatálnymi úmrtiami s vrodenými chybami - 29 %, predčasne narodenými deťmi s vrodenými chybami - 17 %, novorodencami s vrodenými chybami - 10 %. , mŕtvo narodené a perinatálne úmrtia - 7 %, predčasné - 2,5 %, všetci novorodenci - 0,7 %.
Väčšina chromozomálnych ochorení je sporadická, vzniká nanovo v dôsledku genomickej (chromozomálnej) mutácie v gaméte zdravého rodiča alebo v prvých deleniach zygoty a nededí sa po generácie, čo súvisí s vysokou úmrtnosťou pacientov v r. predreprodukčné obdobie. Fenotypovým základom chromozomálnych ochorení sú poruchy včasného embryonálneho vývoja. Preto sa patologické zmeny vyvíjajú už v prenatálnom období vývoja organizmu a buď spôsobujú smrť embrya alebo plodu, alebo vytvárajú hlavné klinický obraz ochorenia už u novorodenca (s výnimkou anomálií pohlavného vývoja, ktoré vznikajú najmä v období puberty). Včasné a viacnásobné poškodenie telesných systémov je charakteristické pre všetky formy chromozomálnych ochorení. Ide o kraniofaciálnu dysmorfiu, vrodené vývojové chyby vnútorné orgány a častí tela, pomalý vnútromaternicový a postnatálny rast a vývoj, mentálna retardácia, poruchy centrálneho nervového systému, kardiovaskulárneho, respiračného, urogenitálneho, tráviaceho a endokrinného systému, ako aj odchýlky hormonálneho, biochemického a imunologického stavu. Každý chromozomálny syndróm je charakterizovaný komplexom vrodených malformácií a vývojových anomálií, ktoré sú do určitej miery vlastné len tomuto typu chromozomálnej patológie. Klinický polymorfizmus každého chromozomálneho ochorenia v jeho všeobecnej forme je určený genotypom organizmu a podmienkami prostredia. Variácie v prejavoch patológie môžu byť veľmi široké - od smrteľného účinku až po menšie vývojové odchýlky. Napriek dobrému štúdiu klinických prejavov a cytogenetiky chromozomálnych ochorení, ich patogenéza, a to ani vo všeobecnosti, nie je zatiaľ jasná. Nevyvinuté všeobecná schéma vývoj zložitých patologických procesov spôsobených chromozomálnymi abnormalitami a vedúcich k vzniku komplexných fenotypov chromozomálnych ochorení.
Hlavné typy chromozomálnych abnormalít
Všetky chromozomálne choroby podľa typu mutácií možno rozdeliť do dvoch veľkých skupín: choroby spôsobené zmenami v počte chromozómov pri zachovaní ich štruktúry (genómové mutácie) a choroby spôsobené zmenami v štruktúre chromozómov (chromozomálne mutácie). Genomické mutácie vznikajú v dôsledku nondisjunkcie alebo straty chromozómov počas gametogenézy alebo v skorých štádiách embryogenézy. U ľudí sa našli iba tri typy genómových mutácií: tetraploidia, triploidia a aneuploidia. Výskyt triploidných (Zn=69) a tetraploidných (4n=92) mutácií je veľmi nízky, nachádzajú sa najmä medzi spontánne potratenými embryami alebo plodmi a u mŕtvo narodených detí. Očakávaná dĺžka života novorodencov s takýmito poruchami je niekoľko dní. Genomické mutácie na jednotlivých chromozómoch sú početné a tvoria väčšinu chromozomálnych ochorení. Okrem toho sa zo všetkých variantov aneuploidie nachádza iba trizómia na autozómoch, polyzómia na pohlavných chromozómoch (tri-, tetra- a pentazómia) a medzi monozómiami sa nachádza iba monozómia X.
Kompletné trizómie alebo monozómie telo znáša ťažšie ako čiastočné, nerovnováha veľkých chromozómov sa u živonarodených detí vyskytuje oveľa menej často ako u malých. Kompletné formy chromozomálnych abnormalít spôsobujú podstatne závažnejšie abnormality ako mozaikové. Autozomálne monozómie sú medzi živonarodenými veľmi zriedkavé, sú to mozaikové formy s veľkým podielom normálnych buniek. Je dokázaná skutočnosť relatívne nízkej genetickej hodnoty heterochromatických oblastí chromozómov. To je dôvod, prečo sú pozorované úplné trizómie u živonarodených detí v tých autozómoch, ktoré sú bohaté na heterochromatín - 8, 9, 13, 14, 18, 21, 22 a X. To vysvetľuje dobrú toleranciu pacientov dokonca aj pri trojitej dávke Y- chromozómový materiál a takmer úplná strata jeho dlhého ramena Úplná monozómia na X chromozóme, kompatibilná s postnatálnym životom, vedúca k rozvoju Shereshevského-Turnerovho syndrómu, ako aj tetra- a pentazómia, sa pozoruje iba na X chromozóme, ktorý je heterochromatický.
Chromozomálne mutácie alebo štrukturálne chromozomálne prestavby sú karyotypové poruchy sprevádzané alebo nesprevádzané nerovnováhou genetického materiálu v rámci jedného alebo viacerých chromozómov (intra- a interchromozomálne preskupenia).
V drvivej väčšine prípadov štrukturálne chromozomálne mutácie prenáša na potomkov jeden z rodičov, ktorého karyotyp obsahuje vyváženú chromozomálnu prestavbu. Patrí medzi ne recipročná (vzájomná) vyvážená translokácia bez straty častí chromozómov, ktoré sa na nej podieľajú. Rovnako ako inverzia nespôsobuje u nosiča patologické javy. Počas tvorby gamét z nosičov vyvážených translokácií a inverzií však môžu vzniknúť nevyvážené gaméty. Robertsonova translokácia - translokácia medzi dvoma akrocentrickými chromozómami so stratou ich krátkych ramien - vedie k vytvoreniu jedného metacentrického chromozómu namiesto dvoch akrocentrických. Nositelia tejto translokácie sú zdraví, pretože strata krátkych ramien dvoch akrocentrických chromozómov je kompenzovaná prácou rovnakých génov vo zvyšných 8 akrocentrických chromozómoch. Počas dozrievania zárodočných buniek náhodné rozdelenie(pri delení buniek) dvoch preskupených chromozómov a ich homológov vedie k vzniku niekoľkých typov gamét, z ktorých niektoré sú normálne, iné obsahujú takú kombináciu chromozómov, ktorá po oplodnení vytvorí zygotu s vyváženým preskupeným karyotypom, pričom iné produkujú počas oplodnenia chromozomálne nevyvážené zygoty.
Pri nevyváženej sade chromozómov (delécie, duplikácie, inzercie) sa u plodu vyvinú závažné klinické patológie, zvyčajne vo forme komplexu vrodených vývojových chýb. Nedostatok genetického materiálu spôsobuje vážnejšie vývojové chyby ako jeho nadbytok.
Oveľa menej často vznikajú štrukturálne aberácie de novo. Rodičia pacienta s chromozomálnou poruchou sú zvyčajne karyotypicky normálni. Chromozomálne ochorenie sa v týchto prípadoch vyskytuje de novo v dôsledku prenosu genómovej alebo chromozomálnej mutácie od jedného z rodičov, ktorá sa vyskytuje raz v jednej z gamét, alebo sa takáto mutácia vyskytuje už v zygote. To nevylučuje recidívu chromozomálnej poruchy u detí v danej rodine. Existujú rodiny predisponované k opakovaným prípadom chromozómovej nondisjunkcie. Mutácie, ktoré vznikli de novo, zodpovedajú za takmer všetky prípady známych úplných trizómií a monozómií. Hlavným mechanizmom výskytu štruktúrnych preskupení akéhokoľvek typu je zlom v jednom alebo viacerých chromozómoch s následným opätovným zjednotením výsledných fragmentov.
Klinické indikácie pre cytogenetickú diagnostiku
Cytogenetická výskumná metóda zaujíma popredné miesto medzi laboratórnymi diagnostickými metódami v medicínskom genetickom poradenstve a prenatálnej diagnostike. Treba sa však striktne držať cieľa
indikácie na odosielanie pacientov na testovanie karyotypu.
Hlavné indikácie prenatálnej diagnostiky:
chromozomálna abnormalita u predchádzajúceho dieťaťa v rodine;
mŕtvo narodené dieťa s chromozomálnou abnormalitou;
chromozomálne preskupenia, chromozomálna mozaika alebo aneuploidia na pohlavných chromozómoch u rodičov;
výsledky testu krvného séra matky naznačujúce zvýšené riziko chromozomálnej abnormality u plodu (riziková skupina);
vek matky;
anomálie plodu zistené ultrazvukovým vyšetrením;
podozrenie na mozaicizmus u plodu počas predchádzajúcej cytogenetickej štúdie;
podozrenie na syndróm chromozomálnej nestability.
Testovanie karyotypu na postnatálnu diagnostiku sa odporúča, ak má pacient:
primárna alebo sekundárna amenorea alebo skorá menopauza;
abnormálny spermogram - azoospermia alebo ťažká oligospermia;
klinicky významné abnormality rastu (nízke, vysoký rast) a veľkosť hlavy (mikro-, makrocefália);
abnormálne genitálie;
abnormálny fenotyp alebo dysmorfia;
vrodené malformácie;
mentálna retardácia alebo vývojové poruchy;
prejavy syndrómu delécie/mikrodelecie/duplikácie;
X-viazané recesívne ochorenie u žien;
klinické prejavy syndrómov chromozomálnej nestability;
pri monitorovaní po transplantácii kostnej drene.
Cytogenetické štúdie by sa mali vykonávať u manželského páru:
s chromozomálnymi abnormalitami resp neobvyklé možnosti chromozómy u plodu zistené počas prenatálnej diagnostiky;
opakované potraty (3 alebo viac); mŕtve narodenie, neonatálna smrť plodu, neschopnosť vyšetriť postihnutý plod;
dieťa má chromozomálnu abnormalitu alebo neobvyklý chromozomálny variant;
neplodnosť neznámej etiológie.
Indikáciou pre cytogenetický výskum je prítomnosť príbuzných pacienta:
chromozomálne preskupenia;
mentálna retardácia pravdepodobne chromozomálneho pôvodu;
reprodukčné straty, vrodené vývojové chyby plodu alebo mŕtve narodenie neznámeho pôvodu.
Indikácie pre výskum metódou FISH:
podozrenie na mikrodelečný syndróm, pre ktorý je dostupná molekulárna cytogenetická diagnostika (dostupnosť vhodných DNA sond);
zvýšené riziko mikrodelečného syndrómu na základe anamnestických údajov;
klinické príznaky naznačujúce mozaicizmus v dôsledku určitého chromozomálneho syndrómu;
stavy po transplantácii kostnej drene, keď darca a príjemca sú rôzneho pohlavia;
podozrenie na chromozomálnu abnormalitu počas štandardnej cytogenetickej štúdie, kedy môže byť metóda FISH užitočná pre ďalšie
objasnenie povahy anomálie alebo v situáciách, keď existujú charakteristické klinické prejavy;
prítomnosť nadpočetného markerového chromozómu;
podozrenie na skrytú chromozomálnu prestavbu.
Metóda FISH na analýzu metafáz je indikovaná:
s markerovými chromozómami;
ďalší materiál neznámeho pôvodu na chromozóme;
chromozomálne preskupenia;
podozrenie na stratu chromozomálneho segmentu;
mozaikovitosť.
Metóda FISH na analýzu interfázových jadier je indikovaná:
s numerickými chromozomálnymi abnormalitami;
duplikáty;
divízie;
chromozómové prestavby;
určenie chromozomálneho pohlavia;
génová amplifikácia.
Cytogenetické výskumné metódy:
Výskum a popis charakteristické znaky metafázové chromozómy sú dôležité najmä pre praktickú cytogenetiku. Jednotlivé chromozómy v rámci skupiny sa rozpoznávajú pomocou techník diferenciálneho farbenia. Tieto metódy umožňujú zistiť heterogenitu chromozómovej štruktúry pozdĺž dĺžky, ktorá je určená charakteristikami komplexu hlavných molekulárnych zložiek chromozómov - DNA a proteínov. Problém rozpoznávania jednotlivých chromozómov v karyotype je dôležitý pre rozvoj cytogenetickej diagnostiky chromozomálnych ochorení u ľudí.
Cytogenetické metódy výskumu sa delia na priame a nepriame. Priame metódy sa používajú v prípadoch, keď je potrebný rýchly výsledok a je možné získať preparáty chromozómov buniek deliacich sa v tele. Medzi nepriame metódy patrí ako povinný krok viac-menej dlhodobá kultivácia buniek v umelých živné médiá. Metódy, ktoré zahŕňajú krátkodobú kultiváciu (od niekoľkých hodín do 2-3 dní), zaujímajú strednú polohu.
Hlavným predmetom cytogenetického výskumu pomocou priamych a nepriamych metód je metafáza mitózy a rôzne štádiá meiózy. Metafáza mitózy je hlavným predmetom cytogenetického výskumu, pretože práve v tomto štádiu je možná presná identifikácia chromozómov a detekcia ich anomálií. Chromozómy v meióze sa skúmajú, aby sa zistili určité typy preskupení, ktoré svojou povahou nie sú detekované v metafáze mitózy.
Biologický materiál pre cytogenetické štúdie. Spracovanie bunkových kultúr. Príprava chromozómových preparátov
Bunky akéhokoľvek tkaniva dostupného na biopsiu možno použiť ako materiál na získanie ľudských chromozómov a ich štúdium. Najčastejšie sa používa periférna krv, kožné fibroblasty, kostná dreň, bunky plodovej vody a bunky choriových klkov. Ľudské lymfocyty periférnej krvi sú najdostupnejšie pre výskum chromozómov.
V súčasnosti takmer všetky laboratóriá na svete používajú metódu využívajúcu na kultiváciu lymfocytov plnú periférnu krv. Krv v množstve 1-2 ml sa odoberie vopred z kubitálnej žily do sterilnej skúmavky alebo fľaštičky s roztokom heparínu. Krv v injekčnej liekovke sa môže uchovávať 24-48 hodín v chladničke pri teplote 4-6 °C. Kultivácia lymfocytov sa uskutočňuje v špeciálnej boxovej miestnosti alebo v pracovná miestnosť pod digestorom s laminárnym prúdením za sterilných podmienok. Takéto podmienky sú povinné, aby sa zabránilo zavlečeniu patogénnej flóry do krvnej kultúry. Pri podozrení na kontamináciu krvi alebo iného materiálu je potrebné do kultivačnej zmesi pridať antibiotiká. Fľaštičky s kultivačnou zmesou sa inkubujú v termostate pri teplote +37 °C počas 72 hodín (prebieha aktívny rast a delenie buniek). Hlavným účelom metodických techník pri spracovaní bunkových kultúr a príprave chromozómových preparátov z nich je získať na preparáte dostatočný počet metafázových doštičiek s takým rozložením chromozómov, aby bolo možné odhadnúť dĺžku, tvar a ďalšie morfologické vlastnosti každého z nich. chromozóm v sade.
K akumulácii buniek v metafáze mitózy a produkcii vysokokvalitných platničiek na prípravku dochádza pomocou niekoľkých sekvenčných postupov:
kolchinizácia - vystavenie buniek cytostatikám kolchicín alebo kolcemid, blokovanie mitózy v štádiu metafázy;
hypotonizácia kultúr;
fixácia buniek zmesou metylalkoholu a kyseliny octovej;
nanesenie bunkovej suspenzie na podložné sklíčko.
Kolchinizácia bunkových kultúr sa uskutočňuje 1,5 až 2 hodiny pred začiatkom fixácie. Po podaní kolchicínu fľaštičky s bunkovou kultúrou pokračujú v inkubácii v termostate. Na konci inkubácie sa kultivačná zmes z každej fľaše naleje do čistých centrifugačných skúmaviek a podrobí sa centrifugácii. Potom sa do bunkového sedimentu pridá hypotonický roztok chloridu draselného, predhriaty na teplotu +37 °C.
Hypotonizácia sa uskutočňuje v termostate pri teplote +37 °C počas 15 minút. Hypotonický roztok KCI podporuje lepšie šírenie chromozómov na podložnom sklíčku. Po hypotonizácii sa bunky sedimentujú centrifugáciou a podrobia sa fixácii. Fixácia sa uskutočňuje zmesou metyl (alebo etyl) alkoholu a kyseliny octovej.
Poslednou fázou je príprava chromozómových preparátov na získanie dobre rozložených metafázových platničiek pri zachovaní integrity a úplnosti chromozómovej sady v každej z nich. Bunková suspenzia sa aplikuje na vlhké, ochladené sklíčka, potom sa sklíčka sušia pri teplote miestnosti a označia sa.
Metódy diferenciálneho farbenia chromozómov
Od roku 1971 sa v cytogenetike rozšírili metódy, ktoré umožňujú diferencovane farbiť každý chromozóm zo sady podľa jeho dĺžky. Praktický význam týchto metód spočíva v tom, že diferenciálne farbenie umožňuje identifikáciu všetkých ľudských chromozómov vďaka špecifickému vzoru pozdĺžneho farbenia pre každý chromozóm. Na farbenie môže byť vhodná akákoľvek farba pozostávajúca zo základného farbiva, pretože hlavným farbiacim substrátom chromozómov je komplex DNA-proteín. V praxi cytogenetického výskumu najväčšie uplatnenie dostali nasledujúce metódy.
Metóda farbenia G je najbežnejšou metódou vďaka svojej jednoduchosti, spoľahlivosti a dostupnosti potrebných činidiel. Každý pár chromozómov po zafarbení získa dĺžkové ryhy v dôsledku striedania rôznofarebných heterochromatických (tmavých) a euchromatických (svetlých) segmentov, ktoré sa zvyčajne označujú ako G-segmenty. Metóda farbenia C poskytuje identifikáciu iba určitých oblastí chromozómov. Ide o oblasti heterochromatínu lokalizované v pericentromérnych oblastiach dlhých ramien chromozómov 1, 9 a 16 a v dlhom ramene chromozómu Y, ako aj v krátkych ramenách akrocentrických chromozómov. R-metóda farbenia chromozómových preparátov ukazuje obraz diferenciálnej segmentácie inverzný k G-metóde. Táto metóda dobre farbí distálne segmenty chromozómov, čo je veľmi dôležité pri identifikácii malých preskupení zahŕňajúcich koncové časti. Metóda farbenia Q poskytuje diferenciálne fluorescenčné farbenie jednotlivých chromozómov súboru, umožňuje identifikovať každý pár homológov a tiež určiť prítomnosť chromozómu Y v interfázových jadrách podľa žiary Y-chromatínového tela.
Princípy chromozómovej analýzy
Povinnou fázou štúdie je vizuálna analýza chromozómov pod mikroskopom s tisícnásobným zväčšením (x1000) s 10x okulármi a 100x imerznou šošovkou. Hodnotenie kvality a vhodnosti chromozómových preparátov na výskum, ako aj výber metafázových platní na analýzu, sa uskutočňuje pri malom zväčšení (x100). Na štúdiu sa vyberú dobre zafarbené, kompletné metafázové platne s dobrým šírením chromozómov. Výskumník spočíta celkový počet chromozómov a posúdi štruktúru každého chromozómu porovnaním pruhovania homológov, ako aj porovnaním pozorovaného vzoru s cytogenetickými mapami (schémami) chromozómov.
Používanie systémov počítačovej analýzy obrazu výrazne zjednodušuje úlohu cytogenetika, skvalitňuje jeho prácu a poskytuje možnosť rýchlo a jednoducho dokumentovať výsledky výskumu. Na zabezpečenie vysokej kvality práce sa odporúča, aby sa na cytogenetickej štúdii každej vzorky zúčastnili dvaja špecialisti. Dokument potvrdzujúci štúdiu je protokol, ktorý uvádza súradnice vyšetrovaných buniek, počet chromozómov v každej z nich, zistené prestavby, vzorec a záver karyotypu, ako aj priezvisko pacienta, dátum a číslo štúdia, priezvisko a podpis lekára (lekárov), ktorí štúdiu viedli . Snímky a obrázky chromozómov by sa mali uložiť na neskoršie preskúmanie.
ZÁKLADNÉ PRAVIDLÁ POPISU CHROMOZOMÁLNYCH ANOMÁLIE PODĽA MEDZINÁRODNÉHO SYSTÉMU CYTOGENETICKEJ NOMENKLATURY
Zaznamenávanie vzorca karyotypu sa musí vykonávať v súlade s aktuálnou verziou Medzinárodného systému pre ľudskú cytogenetickú nomenklatúru. Nižšie uvažujeme o aspektoch používania nomenklatúry, s ktorými sa najčastejšie stretávame v klinickej cytogenetickej praxi.
Počet a morfológia chromozómov:
V karyotype sú chromozómy rozdelené do siedmich ľahko rozlíšiteľných skupín (A-G) podľa ich veľkosti a polohy centroméry. Autozómy sú chromozómy 1 až 22, pohlavné chromozómy sú X a Y.
Skupina A (1-3) - veľké metacentrické chromozómy, ktoré sa dajú od seba odlíšiť veľkosťou a polohou centroméry.
Skupina B (4-5) - veľké submetacentrické chromozómy.
Skupina C (6-12, X) - metacentrické a submetacentrické chromozómy strednej veľkosti. Chromozóm X je jedným z najväčších chromozómov v tejto skupine.
Skupina D (13-15) - stredne veľké akrocentrické chromozómy so satelitmi.
Skupina E (16-18) - relatívne malé metacentrické a submetacentrické chromozómy.
Skupina F (19-20) - malé metacentrické chromozómy.
Skupina G (21-22, Y) - malé akrocentrické chromozómy so satelitmi. Chromozóm Y nemá žiadne satelity.
Každý chromozóm pozostáva z kontinuálnej série pruhov, ktoré sú umiestnené pozdĺž dĺžky chromozómových ramien v prísne ohraničených oblastiach (rezoch). Chromozomálne oblasti sú špecifické pre každý chromozóm a sú nevyhnutné na ich identifikáciu. Pásy a oblasti sú očíslované v smere od centroméry k telomére pozdĺž dĺžky každého ramena. Oblasti sú úseky chromozómu umiestnené medzi dvoma susednými pásmi. Na označenie krátkeho a dlhého ramena chromozómov sa používajú tieto symboly: p - krátke rameno a q - dlhé rameno. Centroméra (sep) je označená symbolom 10, časť centroméry priliehajúca ku krátkemu ramenu je p10 a k dlhému ramenu je q10. Oblasť najbližšie k centromére je označená číslom 1, ďalšia oblasť číslom 2 atď.
Štvorciferná symbolika sa používa na označenie chromozómov:
1. znak - číslo chromozómu;
2. znak (p alebo q) - rameno chromozómu;
3. znak - číslo okresu (sekcie);
4. znak je číslo jazdného pruhu v tejto oblasti.
Napríklad záznam 1p31 označuje chromozóm 1, jeho krátke rameno, oblasť 3, pás 1. Ak je pás rozdelený na subpásy, za označením pásma sa umiestni bodka, potom sa zapíše číslo každého subpásu. Čiastkové pásy, podobne ako pruhy, sú očíslované v smere od centroméry k telomére. Napríklad v pásme 1p31 sú tri subpásy: 1p31.1, 1p31.2 a 1p31.3, z ktorých subpásmo 1p31.1 je proximálne od centroméry a subpásmo 1p31.3 je distálne. Ak sa čiastkové pásma ďalej delia na časti, sú očíslované číslami bez interpunkcie. Napríklad subpásmo 1R31.1 je rozdelené na 1R31.11, 1R31.12 atď.
VŠEOBECNÉ ZÁSADY POPISU NORMÁLNEHO A ABNORMÁLNEHO KARIOTYPU
V popise karyotypu prvý bod označuje celkový počet chromozómov vrátane pohlavných chromozómov. Prvé číslo sa od zvyšku záznamu oddelí čiarkou, potom sa zapíšu pohlavné chromozómy. Autozómy sú určené iba v prípadoch abnormalít.
Normálny ľudský karyotyp vyzerá takto:
46,XX - normálny karyotyp ženy;
46, XY je normálny karyotyp muža.
V prípade chromozomálnych anomálií sa najprv zaznamenávajú anomálie pohlavných chromozómov, potom autozomálne anomálie vo vzostupnom poradí čísel a bez ohľadu na typ anomálie. Každá anomália je oddelená čiarkou. Písmenové označenia sa používajú na opis štrukturálne preskupených chromozómov. Chromozóm, ktorý sa podieľa na prestavbe, sa píše v zátvorkách za symbol označujúci typ preskupenia, napríklad: inv(2), del(4), r(18). Ak sa na preskupení podieľajú dva alebo viac chromozómov, medzi číslo každého chromozómu sa umiestni bodkočiarka (;).
Značky (+) alebo (-) sú umiestnené pred chromozómom, aby indikovali abnormalitu, čo znamená ďalší alebo chýbajúci chromozóm (normálny alebo abnormálny), napríklad: +21,-7,+der(2). Používajú sa aj na označenie zmenšenia alebo zväčšenia dĺžky ramena chromozómu za symbolom (p alebo q); na tento účel možno vyššie uvedené znaky použiť len v texte, nie však v popise karyotypu, napr.: 4p+, 5q-. Pri popise veľkostí heterochromatických segmentov, satelitov a satelitných vlákien sa znamienko (+) (zvýšenie) alebo (-) (zníženie) umiestni bezprostredne za označenie príslušného symbolu, napríklad: 16qh+, 21ps+, 22pstk+. Znak násobenia (x) sa používa na opis viacerých kópií preskupených chromozómov, ale nemožno ho použiť na opis viacerých kópií normálnych chromozómov, napríklad: 46,XX,del(6)(q13q23)x2. Na označenie alternatívnych interpretácií anomálií použite symbol (alebo), napríklad: 46,XX,del(8)(q21.1) alebo i(8)(p10).
Karyotypy rôznych klonov sú oddelené lomkou (/). Hranaté zátvorky sú umiestnené za popisom karyotypu na označenie absolútneho počtu buniek v danom klone. Na označenie dôvodu vzniku rôznych klonov sa používajú symboly mos (mozaika - bunkové línie pochádzajúce z rovnakej zygoty) a chi (chiméra - bunkové línie pochádzajúce z rôznych zygót), ktoré sú uvedené pred popisom zygoty. karyotyp. Pri uvádzaní karyotypov je normálny diploidný klon vždy uvedený ako posledný, napríklad: mos47,XY,+21/46,XY; mos47,XXY/46,XY.
Ak existuje niekoľko anomálnych klonov, záznam sa vykoná v poradí narastajúcej veľkosti: prvý je najčastejšie sa vyskytujúci, potom zostupný. Posledný je normálny klon, napríklad: mos45,X/47,XXX/46,XX. Podobný zápis sa používa v karyotype, ktorý má dva normálne klony, napríklad: chi46,XX/46,XY. Ak sú v karyotype prítomné dva anomálne klony, z ktorých jeden má numerickú anomáliu a druhý má štrukturálne preskupenie, potom sa najprv zaznamená klon s numerickou anomáliou. Napríklad: 45,X/46,X,i(X)(q10).
Keď majú obidva klony číselné anomálie, najskôr sa zaznamená klon s autozómom s nižším poradovým číslom, napríklad: 47,XX,+8/47,XX,+21; klon s abnormalitami pohlavných chromozómov je vždy umiestnený na prvom mieste, napríklad: 47,ХХХ/47,ХХ,+21.
To, že karyotyp je haploidný alebo polyploidný, bude zrejmé z počtu chromozómov a ďalších označení, napr.: 69,XXY. Všetky zmenené chromozómy musia byť označené vo vzťahu k príslušnej úrovni ploidie, napríklad: 70,XXY,+21.
Materský alebo otcovský pôvod abnormálneho chromozómu je označený symbolmi mat a pat po opísanej anomálii, napríklad: 46,XX,t(5;6)(q34;q23)mat,inv(14)( q12q31)pat; 46,XX,t(5;6)(q34;q23)mat,inv(14) (q12q31)mat. Ak je známe, že chromozómy rodičov sú v porovnaní s danou anomáliou normálne, považuje sa za nový a označí sa symbolom denovo (dn), napríklad: 46,XY,t(5;6)(q34 ;q23)mat,inv (14)( q12q31)dn.
Popis numerických chromozómových abnormalít:
Znamienko (+) alebo (-) sa používa na označenie straty alebo získania ďalšieho chromozómu pri popise numerických anomálií.
47,XX,+21 - karyotyp s trizómiou 21.
48,XX,+13,+21 - karyotyp s trizómiou 13 a trizómiou 21.
45,XX,-22 - karyotyp s monozómiou 22.
46,XX,+8,-21 - karyotyp s trizómiou 8 a monozómiou 21.
Výnimkou z tohto pravidla sú ústavné abnormality pohlavných chromozómov, ktoré sa zapisujú bez použitia znamienok (+) a (-).
45,X - karyotyp s jedným X chromozómom (Shereshevsky-Turnerov syndróm).
47,XXY - karyotyp s dvoma X chromozómami a jedným Y chromozómom (Klinefelterov syndróm).
47,XXX - karyotyp s tromi X chromozómami.
47,XYY - karyotyp s jedným X chromozómom a dvoma Y chromozómami.
48,XXXY je karyotyp s tromi chromozómami X a jedným chromozómom Y.
Popis štrukturálnych abnormalít chromozómov
Pri popise štrukturálnych zmien sa používajú systémy krátkeho aj podrobného záznamu. Pri použití krátkeho systému sa uvádza len typ chromozomálnej prestavby a body zlomu. Zapíšte si typ chromozomálnej abnormality, chromozóm zapojený do abnormality a hraničné body v zátvorkách. Krátky systém neumožňuje jednoznačný popis zložitých chromozomálnych prestavieb, ktoré sa niekedy zisťujú pri analýze karyotypov nádorov.
Stručný systém označovania konštrukčných úprav
Ak sú obe ramená zapojené do prestavby, ktorá je výsledkom dvoch zlomov vyskytujúcich sa na jednom chromozóme, bod zlomu v krátkom ramene sa zaznamená pred bodom zlomu v dlhom ramene: 46,XX,inv(2)(p21q31). Keď sú dva body zlomu na tom istom ramene chromozómu, bod zlomu proximálne od centroméry je označený ako prvý: 46,XX,inv(2)(p13p23). V prípade, že sa na preskupení podieľajú dva chromozómy, ako prvý sa uvádza buď chromozóm s nižším poradovým číslom alebo pohlavný chromozóm: 46,XY,t(12;16)(q13;p11.1); 46, X, t (X; 18) (p11.11; q11.11).
Výnimkou z pravidla sú prestavby s tromi bodmi zlomu, keď sa fragment jedného chromozómu vloží do oblasti iného chromozómu. V tomto prípade je chromozóm príjemcu zapísaný ako prvý a chromozóm darcu ako posledný, aj keď ide o pohlavný chromozóm alebo chromozóm s nižším poradovým číslom: 46,X,ins(5;X)(p14;q21q25); 46,XY,ins(5;2)(p14;q22q32). Ak preskupenie zasiahne jeden chromozóm, najskôr sú označené body zlomu v segmente, kde sa vytvorila inzercia. V prípade priamej inzercie sa najskôr zaznamená bod zlomu vloženého fragmentu proximálne od centroméry a potom bod zlomu distálneho. Pri obrátenom vkladaní je opak pravdou.
Na označenie translokácií, na ktorých sa podieľajú tri rôzne chromozómy, sa najprv uvedie pohlavný chromozóm alebo chromozóm s nižším poradovým číslom, potom chromozóm, ktorý prijal fragment z prvého chromozómu, a nakoniec chromozóm, ktorý fragment daroval prvý chromozóm. 46,XX,t(9;22;17) (q34;q11.2;q22) - fragment chromozómu 9, zodpovedajúci distálnej oblasti 9q34, prenesený na chromozóm 22, na segment 22q11.2, fragment chromozómu 22, zodpovedajúci distálnej oblasti 22q11 .2, sa prenesie na chromozóm 17 v segmente 17q22 a fragment chromozómu 17, zodpovedajúci distálnej oblasti 17q22, sa prenesie na chromozóm 9 v segmente 9q34.
Podrobný systém označovania štrukturálnych zmien. V súlade s podrobným systémom notácie sú štrukturálne prestavby chromozómov určené zložením pásov v nich. Všetky označenia používané v krátky systém, sú uložené aj v podrobnom systéme. Avšak v podrobnom systéme dávajú Detailný popis zloženie pásov v preskupených chromozómoch pomocou ďalších symbolov. Dvojbodka (:) označuje bod zlomu a dvojitá dvojbodka (::) označuje prerušenie, po ktorom nasleduje opätovné spojenie. Šípka (->) označuje smer prenosu chromozómových fragmentov. Konce chromozómových ramien sú označené symbolom ter (terminál), pter alebo qter označujúce koniec krátkeho alebo dlhého ramienka. Symbol sep sa používa na označenie centroméry.
Typy chromozomálnych prestavieb
Ďalší materiál neznámeho pôvodu. Symbol add (z latinského additio - sčítanie) sa používa na označenie ďalšieho materiálu neznámeho pôvodu, ktorý bol pridaný do chromozomálnej oblasti alebo pásu. Ďalší materiál pripojený k terminálnej oblasti spôsobí zväčšenie dĺžky ramena chromozómu. Pri opise chromozómov s doplnkový materiál neznámeho pôvodu v oboch ramenách symbol der je umiestnený pred číslom chromozómu. Ak sa do ramena chromozómu vloží neznámy extra materiál, na popis sa použijú symboly ins a (?).
vymazania. Symbol del sa používa na označenie terminálnych a intersticiálnych vymazaní:
46,XX,del(5)(q13)
46,XX,del (5) (pter->q13:)
Znamienko (:) znamená, že k prerušeniu došlo v pásme 5q13, v dôsledku čoho sa chromozóm 5 skladá z krátkeho ramena a časti dlhého ramena, ktoré sa nachádzajú medzi centromérou a segmentom 5q13.
46,XX,del(5)(q13q33)
46,XX,del(5)(pter->q13::q33->qter)
Znak (::) znamená prerušenie a opätovné spojenie pásov 5ql3 a 5q33 dlhého ramena chromozómu 5. Chromozómový segment medzi týmito pásmi je vymazaný.
Derivátové alebo odvodené chromozómy (der) sú chromozómy, ktoré vznikajú v dôsledku preskupení ovplyvňujúcich dva alebo viac chromozómov, ako aj v dôsledku viacerých preskupení v rámci jedného chromozómu. Počet derivovaných chromozómov zodpovedá číslu intaktného chromozómu, ktorý má rovnakú centroméru ako derivačný chromozóm:
46,XY,der(9)del(9)(p12)del(9)(q31)
46,XY,der(9) (:р12->q31:)
Derivačný chromozóm 9 je výsledkom dvoch terminálnych delécií vyskytujúcich sa v krátkych a dlhých ramenách s bodmi zlomu v pásoch 9p12 a 9q31, v tomto poradí.
46,XX,der (5)add(5)(p15.1)del(5)(q13)
46,XX,der(5)(?::p15.1-»q13:)
Odvodený chromozóm 5 s ďalším materiálom neznámeho pôvodu pripojeným k pásu 5p15.1 a terminálnou deléciou dlhého ramena distálne k pásu 5q13.
Dicentrické chromozómy. Symbol die sa používa na opis dicentrických chromozómov. Dicentrický chromozóm nahrádza jeden alebo dva normálne chromozómy. Nie je teda potrebné indikovať chýbajúce normálne chromozómy.
45,XX,dic(13;13)(q14;q32)
45,XX,dic(13;13)(13pter->13ql4::13q32-»13pter)
K zlomeniu a opätovnému spojeniu došlo v pásoch 13ql4 a 13q32 na dvoch homológnych chromozómoch 13, čo viedlo k dicentrickému chromozómu.
Duplikácie. Duplikácie sú označené symbolom dup; môžu byť priame alebo obrátené.
46,XX,dup(1) (q22q25)
46,XX,dup(1)(pter->q25::q22->qter)
Priama duplikácia segmentu medzi pásmami lq22 a lq25.
46,XY,dup(1)(q25q22)
46,XY,dup(1) (pter->q25::q25->q22::q25->qter) alebo (pter->q22::q25-»q22::q22->qter)
Obrátená duplikácia segmentu medzi pásmami lq22 a lq25. Treba poznamenať, že iba podrobný systém umožňuje popísať obrátenú duplikáciu.
Inverzie. Symbol inv sa používa na opis para- a pericentrických inverzií.
46,XX,inv(3) (q21q26.2)
46,XX,inv(3)(pter->q21::q26.2->q21::q26.2->qter)
Paracentrická inverzia, pri ktorej došlo k zlomu a opätovnému spojeniu v pásoch 3q21 a 3q26.2 dlhého ramena chromozómu 3.
46,XY,inv(3)(p13q21)
46,XY,inv(3)(pter-»pl3::q21->p13::q21->qter)
Pericentrická inverzia, pri ktorej došlo k pretrhnutiu a opätovnému spojeniu medzi pásikom krátkeho ramena 3p13 a pásom dlhého ramena 3q21 chromozómu 3. Oblasť medzi týmito pásmi, vrátane centroméry, je obrátená o 180°.
Vložky. Symbol ins sa používa na označenie priameho alebo obráteného vkladania. Vloženie sa považuje za priame, keď je proximálny koniec oblasti vkladania v proximálnej polohe vzhľadom na jej druhý koniec. Pri obrátenej inzercii je proximálny koniec oblasti vkladania v distálnej polohe. Typ vkladania (priame alebo obrátené) možno tiež označiť symbolmi dir a inv.
46,XX,ins(2) (pl3q21q31)
46,XX,ins(2)(pter->p13::q31->q21::pl3-»q21::q31-qter)
K priamej inzercii, t.j. dir ins(2) (p13q21q31), došlo medzi segmentmi 2q21 a 2q31 dlhého ramena a segmentom 2p13 krátkeho ramena chromozómu 2. Oblasť chromozómu dlhého ramena medzi segmenty 2q21 a 2q31 je vložená do krátke rameno v oblasti segmentu 2p13. V novej polohe zostáva segment 2q21 bližšie k centromére ako segment 2q31.
46,XY,ins(2) (pl3q31q21)
46,XY,ins(2)(pterH>pl3::q21->q31::pl3->q21::q31-»qter)
V tomto prípade je vložená sekcia invertovaná, t.j. inv ins(2)(p13q31q21). Vo vložke je segment 2q21 ďalej od centroméry ako segment 2q31. Zmenilo sa teda umiestnenie segmentov vzhľadom na centroméru.
izochromozómy. Symbol i sa používa na opis izochromozómov, čo sú chromozómy pozostávajúce z dvoch rovnakých ramien. Body zlomu v izochromozómoch sú lokalizované v centromerických oblastiach p10 a q10.
46,XX,i(17)(q10)
46,XX,i(17)(qter-»q10::q10 ->qter)
Izochromozóm pozdĺž dlhého ramena chromozómu 17 a bod zlomu sú označené v oblasti 17q10. Karyotyp obsahuje jeden normálny chromozóm a jeden preskupený chromozóm 17.
46,X,i(X)(q10)
46,X,i(X) (qter-»q10::q10->qter)
Jeden normálny chromozóm X a izochromozóm X pozdĺž dlhého ramena.
Krehké miesta (fragilné miesta) sa môžu javiť ako normálne polymorfizmy alebo môžu byť spojené s dedičnými chorobami alebo fenotypovými abnormalitami.
46,X,fra(X)(q27,3)
Krehká oblasť v subpásme Xq27.3 jedného z chromozómov X v ženskom karyotype.
46,Y,fra(X)(q27,3)
Krehká oblasť v subpásme Xq27.3 chromozómu X v mužskom karyotype.
Markerový chromozóm (značka) je štrukturálne zmenený chromozóm, ktorého časť nemožno identifikovať. Ak je identifikovaná akákoľvek časť abnormálneho chromozómu, je opísaná ako odvodený chromozóm (der). Pri popise karyotypu sa pred symbol mar umiestni znamienko (+).
47,XX,+mar
Jeden ďalší markerový chromozóm.
48,X,t(X;18)(p11.2;q11.2)+2mar
Dva markerové chromozómy navyše k translokácii t(X;18).
Kruhové chromozómy sú označené symbolom r a môžu pozostávať z jedného alebo viacerých chromozómov.
46,XX,r(7)(p22q36)
46,XX,r(7) (::р22->q36::)
K zlomeniu a opätovnému zjednoteniu došlo v segmentoch 7p22 a 7q36 so stratou chromozómových oblastí distálnych od týchto zlomových bodov.
Ak centroméra kruhového chromozómu nie je známa, ale segmenty chromozómov obsiahnuté v kruhu sú známe, kruhové chromozómy sú definované ako deriváty (der).
46,XX,der(1)r(1;3)(p36.1q23;q21q27)
46,XX,der(1)(::lp36.1->1q23::3q21->3q27::)
Premiestnenia. Recipročné translokácie
Na opis translokácií (t) sa používajú rovnaké princípy a pravidlá ako na opis iných chromozomálnych prestavieb. Na rozlíšenie homológnych chromozómov môže byť jeden z homológov podčiarknutý jedným podčiarkovníkom (_).
46,XY,t(2;5)(q21;q31)
46,XY,t(2;5)(2pter2q21::5q31->5qter;5pter 5q31::2q21->2qter)
Prestávka a opätovné spojenie nastalo v segmentoch 2q21 a 5q31. Chromozómy si vymenili oblasti distálne od týchto segmentov. Chromozóm s nižším sériovým číslom je označený ako prvý.
46,X,t(X;13)(q27;ql2)
46,X,t(X;13)(Xpter->Xq27::13ql2->13qter;13pter->3q 12::Xq27->Xqter)
Prestávka a opätovné spojenie nastali v segmentoch Xq27 a 13q12. Segmenty distálne od týchto oblastí boli vymenené. Keďže sa na translokácii podieľa pohlavný chromozóm, zaznamenáva sa ako prvý. Všimnite si, že správny zápis je 46,X,t(X;13), nie 46,XX,t(X;13).
46,t(X;Y) (q22;ql, 1,2)
46,t(X;Y)(Xpter->Xq22::Yq11.2->Yqter;Ypter->Yq11.2::Xq22->Xqter)
Recipročná translokácia medzi chromozómami X a Y s bodmi zlomu Xq22 a Yq11.2.
Translokácie zahŕňajúce celé ramená chromozómov možno zaznamenať, čo naznačuje zlomové body v centromerických oblastiach p10 a q10. Pri vyvážených translokáciách je bod zlomu v pohlavnom chromozóme alebo v chromozóme s nižším poradovým číslom označený ako p10.
46,XY,t(4;3)(p10;q10)
46,XY,t(1;3)(lpteMlpl0::3ql0->3qter;3pter->3p40::4q40->4qter)
Recipročná translokácia celých ramien chromozómu, pri ktorej sa krátke ramená chromozómu 1 spojili s centromérou s dlhými ramenami chromozómu 3 a dlhé ramená chromozómu 1 sa spojili s krátkymi ramenami chromozómu 3.
Pri nevyvážených translokáciách celých chromozómových ramien je preusporiadaný chromozóm označený ako derivát (der) a nahrádza dva normálne chromozómy.
45,XX,der(1;3) (p10;q10)
45,XX,der(1;3)(1pter->1p10::3q10->3qter)
Odvodený chromozóm pozostávajúci z krátkeho ramena chromozómu 1 a dlhého ramena chromozómu 3. Chýbajúce chromozómy 1 a 3 nie sú označené, pretože sú nahradené odvodeným chromozómom. Karyotyp teda obsahuje jeden normálny chromozóm 1, jeden normálny chromozóm 3 a odvodený chromozóm der(l;3).
Robertsonove translokácie
Ide o špeciálny typ translokácie, ktorý vzniká v dôsledku centrickej fúzie dlhých ramien akrocentrických chromozómov 13-15 a 21-22 so súčasnou stratou krátkych ramien týchto chromozómov. Princípy opisu nevyvážených translokácií zahŕňajúcich celé ramená platia aj pre opis Robertsonových translokácií pomocou symbolu (der). Symbol rob možno použiť aj na opis týchto translokácií, ale nemal by sa používať na opis získaných anomálií. Body zlomu chromozómov zapojených do translokácie sú uvedené v oblastiach q10.
45,XX,der(13;21) (q10;q10)
45,XX,rob(13;21) (q10;q10)
K zlomeniu a opätovnému spojeniu došlo v segmentoch 13q10 a 21q10 centromerických oblastí chromozómov 13 a 21. Odvodený chromozóm nahradil jeden chromozóm 13 a jeden chromozóm 21. Chýbajúce chromozómy nie je potrebné uvádzať. Karyotyp obsahuje jeden normálny chromozóm 13, jeden normálny chromozóm 21 a der (13;21). Nerovnováha nastáva v dôsledku straty krátkych ramien chromozómov 13 a 21.
Rozlišujú sa tieto typy mutácií:
A) genómové mutácie,čo vedie k zmene počtu chromozómov. V rastlinách sa často vyskytujú genómové mutácie. V tomto prípade môže dôjsť k zmnoženiu celých sád chromozómov (polyploidia) alebo k zvýšeniu (trizómia) či zníženiu (monozómia) počtu jednotlivých chromozómov;
b) chromozomálne mutácie(pozri časť 2.2), pri ktorých je narušená štruktúra chromozómov, ale ich počet v bunke zostáva nezmenený. Chromozomálne mutácie sa dajú zistiť mikroskopickým vyšetrením.
V) génové mutácie, nevedie k zmenám v chromozómoch, ktoré je možné detegovať pomocou mikroskopu; tieto mutácie možno zistiť len genetickou analýzou fenotypových zmien (pozri časť 3.6).
Štúdium mutácií u ľudí na úrovni bielkovín a DNA (najmä mutácie hemoglobínových génov) výrazne prispelo k pochopeniu ich molekulárnej podstaty. Výsledky týchto štúdií a výsledky analýzy chromozómovej štruktúry pomocou metód diferenciálneho farbenia s vysokým rozlíšením viedli k rozmazaniu hranice medzi chromozomálnymi a génovými mutáciami. Teraz vieme, že delécie a inzercie sú možné na molekulárnej úrovni a že nerovnomerné kríženie môže zmeniť mikroštruktúru. Diferenciálne metódy farbenia umožnili pod mikroskopom odhaliť predtým nerozoznateľné chromozomálne preskupenia. Malo by sa pamätať na to, že chromozomálne zmeny zistené diferenciálnym farbením sa líšia o niekoľko rádov
5 mutácií 143
zo zmien, ako sú delécie štrukturálnych génov. Preto je rozlíšenie medzi štrukturálnymi chromozomálnymi aberáciami a génovými mutáciami užitočné na praktické účely.
Bunky, v ktorých sa vyskytujú mutácie. Okrem typu genetické poškodenie, je mimoriadne dôležité lokalizácia. Mutácie sa môžu vyskytnúť v zárodočných aj somatických bunkách. Tie, ktoré vznikajú v zárodočných bunkách, sa prenášajú na jedincov ďalšej generácie a spravidla sa nachádzajú vo všetkých bunkách potomkov, ktorí sa stávajú ich nosičmi. Somatické mutácie možno detegovať iba v potomstve zodpovedajúcej mutantnej bunky, čo vedie k „mozaikovému“ jedincovi. Fenotypové dôsledky sa objavia iba vtedy, ak tieto mutácie interferujú s implementáciou špecifických funkcií, ktoré sú týmto mutantným bunkám vlastné.
Frekvencie mutácií. Jedným z najčastejšie používaných parametrov pri štúdiu procesu mutácie je frekvencia vznik mutácie(alebo rýchlosť mutácie). Vo vzťahu k človeku je definovaný ako pravdepodobnosť výskytu mutácie počas života jednej generácie. Spravidla sa to týka frekvencie mutácií v oplodnených vajíčkach. Otázka rýchlosti mutácií v somatických bunkách je diskutovaná v časti. 5 1.6.