Mutații de novo. Noile mutații (De Novo) pot fi detectate în embrionii fertilizați folosind diagnosticul genetic preimplantare (PGD). Caracteristicile clinice ale sindromului Noonan
Amniocenteza - un test folosit pentru a obține o probă pentru analiza genelor și cromozomilor fătului. Fătul se află în uter înconjurat de lichid. Acest lichid conține un număr mic de celule ale pielii de la copilul nenăscut. O cantitate mică de lichid este retrasă cu un ac subțire prin peretele abdominal (burta) al mamei. Lichidul este trimis la laborator pentru testare. Pentru mai multe informații, consultați broșura Amniocenteză.
Tulburare genetică autosomal dominantă este o boală pentru care o persoană trebuie să moștenească o copie alterată a unei gene (mutație) de la unul dintre părinții săi. Cu acest tip de moștenire, boala se transmite la jumătate dintre copiii unui cuplu de la unul dintre părinți care este bolnav. Ambele sexe sunt la fel de probabil să fie afectate. În familii există transmisie verticală boli: de la un părinte la jumătate dintre copii.
Genetică autozomal recesivboala - Aceasta este o boală pentru care o persoană trebuie să moștenească două copii modificate ale unei gene (mutație), câte una de la fiecare părinte. Cu acest tip de moștenire, un sfert dintre copiii unui cuplu sunt bolnavi. Părinții sunt sănătoși, dar sunt purtători ai bolii. O persoană care are o singură copie a genei modificate va fi un purtător sănătos. Pentru mai multe informații, consultați broșura Moștenire recesiva.
autozomal - o trăsătură a cărei genă este localizată pe autozomi.
autozomi - Oamenii au 23 de perechi de cromozomi. Perechile de la 1 la 22 se numesc autozomi și arată la fel la bărbați și femei. Cromozomii celei de-a 23-a perechi sunt diferiți la bărbați și la femei și se numesc cromozomi sexuali.
Biopsie vilozități coriale, FVP - o procedură efectuată în timpul sarcinii pentru a colecta celule de la făt pentru a testa genele sau cromozomii copilului nenăscut pentru anumite afecțiuni moștenite. Un număr mic de celule sunt prelevate din placenta în curs de dezvoltare și trimise la un laborator pentru testare. Pentru mai multe informații, consultați broșura Biopsia vilozităților coriale.
vagin - organul care leagă uterul cu mediul extern, canalul de naștere.
gena - informatii necesare organismului sa functioneze, stocate in formă chimică(ADN) pe cromozomi.
genetice - cauzate de gene, legate de gene.
cercetare genetica - un test care poate ajuta la determinarea dacă există modificări în gene sau cromozomi individuali. Pentru mai multe informații, consultați broșura Ce este testarea genetică?
Boala genetica - o boală cauzată de anomalii ale genelor sau cromozomilor.
Ștergere - pierderea unei părți a materialului genetic (ADN); termenul poate fi folosit pentru a se referi la pierderea unei părți atât a unei gene, cât și a unui cromozom. Pentru mai multe informații, consultați broșura Tulburări cromozomiale.
ADN - substanta chimica din care sunt compuse genele si care contine informatiile necesare functionarii organismului.
Duplicare - o repetare anormală a unei secvențe de material genetic (ADN) într-o genă sau cromozom. Pentru mai multe informații, consultați broșura Tulburări cromozomiale.
Măsurarea grosimii spațiului gulerului (TVP) - Examinarea cu ultrasunete a spatelui zonei gâtului fetal, care este umplut cu lichid la începutul sarcinii. Dacă copilul are o afecțiune congenitală (de exemplu, sindromul Down), grosimea translucidității nucale poate fi modificată.
inversare - modificarea secvenței genelor pe un singur cromozom. Pentru mai multe informații, consultați broșura Tulburări cromozomiale.
Inserare - inserarea de material genetic suplimentar (ADN) într-o genă sau cromozom. Pentru mai multe informații, consultați broșura Tulburări cromozomiale.
Cariotip - o descriere a structurii cromozomilor unui individ, inclusiv numărul de cromozomi, numărul de cromozomi sexuali (XX sau XY) și orice abateri de la numărul normal.
Celulă- Corpul uman este format din milioane de celule care servesc" blocuri de construcție" Celulele din diferite locuri ale corpului uman arată diferit și îndeplinesc funcții diferite. Fiecare celulă (cu excepția ovulelor la femei și a spermei la bărbați) conține două copii ale fiecărei gene.
Cromozom inel este un termen folosit atunci când capetele unui cromozom se unesc pentru a forma un inel. Pentru mai multe informații, consultați broșura Translocații cromozomiale.
uterul - partea a corpului femeii în care fătul crește în timpul sarcinii.
Consiliere genetică medicală- informarea si asistenta medicala persoanelor preocupate de prezenta unei afectiuni in familie, eventual de natura ereditara.
Mutaţie- o modificare a secvenței ADN a unei anumite gene. Această modificare a secvenței unei gene duce la faptul că informațiile conținute în ea sunt perturbate și nu pot funcționa corect. Acest lucru poate duce la dezvoltarea unei boli genetice.
Avort spontan - pÎntreruperea prematură a sarcinii care are loc înainte ca bebelușul să poată supraviețui în afara uterului.
Translocare dezechilibrata - translocare, în care o rearanjare cromozomială duce la achiziționarea sau pierderea unei anumite cantități de material cromozomial (ADN), sau simultan la achiziționarea suplimentară și la pierderea unei părți din materialul original. Poate apărea la un copil al cărui părinte este purtător al unei translocații echilibrate. Pentru mai multe informații, consultați broșura Translocații cromozomiale.
Purtător de rearanjare cromozomială - o persoană care are o translocație echilibrată, în care cantitatea de material cromozomial nu este nici redusă, nici crescută, ceea ce de obicei nu provoacă probleme de sănătate.
Transportator - o persoană care nu are de obicei o boală (în prezent), dar este purtătoarea unei copii modificate a unei gene. În cazul unei boli recesive, purtătorul este de obicei sănătos; în cazul unei boli dominante cu debut tardiv, persoana se va îmbolnăvi mai târziu.
Fertilizarea - fuziunea unui ovul și a unui spermatozoid pentru a crea prima celulă a unui copil.
Placenta- un organ adiacent peretelui interior al uterului unei femei gravide. Fătul primește nutrienți prin placentă. Placenta crește dintr-un ovul fertilizat, deci conține aceleași gene ca și fătul.
rezultat pozitiv - un rezultat al testului care arată că persoana testată are o modificare (mutație) într-o genă.
cromozomi sexuali - cromozomul X și cromozomul Y. Setul de cromozomi sexuali determină dacă un individ este bărbat sau femeie. Femeile au doi cromozomi X, bărbații au un cromozom X și un cromozom Y.
Testare predictivă - testare genetică pentru a identifica o afecțiune care se poate dezvolta sau se va dezvolta în timpul vieții. Atunci când un test genetic are ca scop identificarea unei afecțiuni care este aproape sigur că se va dezvolta în viitor, testul este numit presimptomatice.
Diagnosticul prenatal- un studiu efectuat în timpul sarcinii pentru a determina prezența sau absența unei boli genetice la copil.
Translocare reciprocă - o translocare care are loc atunci când două fragmente se desprind din doi cromozomi diferiți și își schimbă locul. Pentru mai multe informații, consultați broșura Translocații cromozomiale.
translocare robertsoniana - apare atunci când un cromozom se atașează de altul. Pentru mai multe informații, consultați broșura Translocații cromozomiale.
Translocare echilibrată - t relocare (rearanjare cromozomială), în care cantitatea de material cromozomial nu este redusă sau crescută, ci este mutată de la un cromozom la altul. O persoană cu o translocare echilibrată de obicei nu suferă de acest lucru, dar riscul de a dezvolta boli genetice pentru copiii săi este crescut. Pentru mai multe informații, consultați broșura Translocații cromozomiale.
Starea aderată la podea- Vezi moștenirea legată de X.
spermatozoizi - celula germinativă a tatălui, contribuția paternă la formarea celulei din care se va dezvolta nou-nascut. Fiecare spermatozoid conține 23 de cromozomi, câte unul din fiecare pereche de cromozomi ai tatălui. Spermatozoizii fuzionează cu ovulul pentru a crea prima celulă din care se dezvoltă copilul nenăscut.
Translocare - rearanjarea materialului cromozomial. Apare atunci când un fragment dintr-un cromozom se rupe și se atașează la o altă locație. Pentru mai multe informații, consultați broșura Translocații cromozomiale.
Examinare cu ultrasunete (ultrasunete) - un test nedureros care folosește unde sonore pentru a crea o imagine a fătului care crește în uterul mamei. Se poate face prin deplasarea capului scanerului de-a lungul suprafeței peretelui abdominal al mamei (stomac) sau în interiorul vaginului.
cromozomi - structuri sub formă de fir vizibile la microscop care conțin gene. În mod normal, o persoană are 46 de cromozomi. Moștenim un set de 23 de cromozomi de la mama noastră, iar al doilea set de 23 de cromozomi de la tatăl nostru.
Boală legată de X- o boală genetică care apare ca urmare a unei mutații (modificări) la o genă situată pe cromozomul X. Bolile legate de X includ hemofilia, distrofia musculară Duchenne, sindromul X fragil și multe altele. Pentru mai multe informații, consultați broșura Moștenire legată de X.
XX- așa este de obicei reprezentat setul de cromozomi sexuali al unei femei. În mod normal, o femeie are doi cromozomi X. Fiecare cromozom X este moștenit de la unul dintre părinți.
cromozomul X - Unul dintre cromozomii sexuali. Femeile au în mod normal doi cromozomi X. Bărbații au în mod normal un cromozom X și un cromozom Y.
Ovar/ovare- organe din corpul unei femei care produc ouă.
ovul - celula reproductivă a mamei, care va servi drept bază pentru crearea primei celule a copilului nenăscut. Oul conține 23 de cromozomi; câte unul din fiecare pereche pe care o are mama. Ovulul fuzionează cu spermatozoizii pentru a forma prima celulă a bebelușului.
De novo - din o expresie latină care înseamnă „din nou”. Folosit pentru a descrie modificări ale genelor sau cromozomilor (mutații) care sunt nou formați, de ex. niciunul dintre părinții unei persoane cu o mutație de novo nu are aceste modificări.
X Y- așa este de obicei reprezentat setul de cromozomi sexuali al unui bărbat. În Noria, bărbații au un cromozom X și un cromozom Y. Bărbații moștenesc cromozomul X de la mama lor și cromozomul Y de la tatăl lor.
cromozomul Y- unul dintre cromozomii sexuali. În mod normal, bărbații au un cromozom Y și un cromozom X. O femeie are în mod normal doi cromozomi X.
Din BrainstormWiki
Ce este o mutație?
Să începem cu faptul că nu există un cod genetic „corect”. Prin urmare, dacă eu am o genă și tu ai alta, asta nu înseamnă că unul dintre noi este un mutant.
Cu toate acestea, există modificări ale codului genetic care duc la probleme evidente, iar acestea sunt de obicei rare; se numesc mutatii. Modificările care apar la mai mult de 1% din populație sunt mai corect numite nu mutații, dar polimorfisme.
Mutațiile pot fi moștenite de la părinți sau pot apărea doar la un copil - caz în care sunt numite de novo
Mutațiile pot apărea în timpul dezvoltării organismului, în timpul diviziunii și diferențierii celulare - acestea se numesc mutatii somatice; nu pot fi moștenite deoarece „trăiesc” în celulele corpului (soma), dar nu și în celulele germinale.
Dimpotrivă, mutațiile care afectează celulele germinale sunt moștenite și sunt numite mutații ale liniei germinale
Mutațiile pot fi tăcut- sunt acolo, dar nu are niciun efect. Acestea sunt marea majoritate - dacă vă amintiți, genele ocupă doar mai puțin de 2% din tot ADN-ul. Fiecare dintre noi este purtător a cel puțin zeci de astfel de mutații. Dacă cad pe secțiuni ADN neimportante, nimeni nu va observa nimic.
Mutațiile pot afecta o „litera” a codului genei - sau pot afecta un întreg fragment uriaș de ADN. Acest fragment poate fi de cele mai multe ori pierdut (ștergere) sau duplicat (duplicare) - în acest caz, celulele vor modifica cantitatea de proteine sintetizate pentru genele afectate - doza acestora se va schimba
Pentru diferite tipuri de mutații există tipuri diferite analize și teste, iar acum vom încerca să le parcurgem.
De la mare la mic
Aneuploidie - o anomalie a numărului de cromozomi
Cea mai mare anomalie genetică este o modificare a numărului de cromozomi. Deși acesta nu este autism, merită să începem cu 50 de ani în urmă s-a descoperit că sindromul Down este cauzat de trei 21 de cromozomi, în loc de doi. Aceasta se numește trisomie; iar o modificare a numărului general de cromozomi este aneuploidia. În plus, există cromozomii trisomiei 13 (sindromul Patau) și 18 (sindromul Edwards), precum și o serie de aneuploidii ale cromozomilor sexuali (X și Y)
Toate acestea pot fi văzute cu un microscop optic obișnuit - o analiză numită „cariotip”. Cromozomii celulelor în diviziune sunt fotografiați, sortați și descriși. Imaginea prezintă cariotipul unei femei cu trei 21 de cromozomi.
Efectul trisomiei este enorm și vizibil în multe procese: în structura corpului, în procesele fiziologice și chiar în proteinele caracteristice din sângele mamei purtătoare (așa funcționează acum testele de screening în satul Dauna)
De ce se găsesc doar trisomiile 21, 13 și 18 și cromozomii sexuali? Acest lucru se poate întâmpla cu orice cromozom, dar restul nu supraviețuiește nici până la stadiul de sarcină vizibilă. Poate că motivul este că cromozomii 21, 13 și 18 sunt printre cei mai săraci în gene proteice (nu uitați, sunt numerotați după mărime, sunt, de asemenea, mici) și creșterea dozei lor este relativ tolerabilă. Acest lucru este confirmat de tabelul de mai jos: în fazele incipiente ale dezvoltării, orice aneuploidie este posibilă, iar pe măsură ce vă apropiați de o naștere reușită, doar aceste 3.

Variația numărului de copiere

CNV este, de asemenea, fie o duplicare, fie absența unei anumite părți a ADN-ului, dar nu la nivelul întregului cromozom, ci a mai multor milioane de nucleotide.
Pentru a le detecta, se folosesc teste numite CGH array: hibridizare genomică comparativă, de asemenea FISH și altele.În practica noastră, acestea sunt adesea numite „cariotipări moleculare”, analiză cromozomială microarray etc.
Acestea sunt încă bucăți uriașe de ADN care pot include zeci de gene și structuri de control. Efectele sunt foarte vizibile și includ aproape întotdeauna dismorfisme și modificări ale abilităților cognitive. Ele sunt desemnate de zona afectată a codului genei, de exemplu ștergerea 2q32 - pierderea unei secțiuni în dunga 32 pe brațul lung 2 cromozomi (amintiți-vă partea 1 și „adresele” secțiunilor cromozomilor) Multe sindroame „numite” binecunoscute aparțin și aici, de exemplu sindromul Williams - ștergerea 7q11.23
Se crede că astfel de mutații CNV explică 3% până la 10% din cazurile de autism. Acesta este practic singurul tip de anomalie genetică care este asociat cu încredere cu unele forme - sindromice - de autism.
Încă o dată, principalul lucru pe care trebuie să-l știi despre CNV-urile în autism: aceștia funcționează într-o proporție redusă de cazuri, dar efectul lor este vizibil în multe aspecte, de la dismorfisme la inteligență. Acestea. prezența defectelor în CNV poate fi observată aproape întotdeauna fie direct pe față, fie sub forma unor tulburări destul de evidente precum hipotensiunea sistemică și ataxia...
Unele CNV-uri cunoscute și documentate

Cu toate acestea, prezența datelor CNV nu garantează autismul ca diagnostic. Penetranța nu este niciodată 100%. Un studiu recent în Estonia asupra unui grup de persoane fără nici un diagnostic a arătat că purtătorii de CNV 16p11.2 prezentau dismorfisme ale capului, obezitate, tulburări cognitive – dar nu aveau un diagnostic de autism, contrar datelor din tabelul de mai sus;
Există o bază de date online cu CNV care provoacă autism și sindroame asociate http://projects.tcag.ca/autism/
Autism sindromic
Autismul sindromic este numit atunci când se crede că simptomele autiste sunt cauzate de o tulburare genetică de înțeles - de obicei CNV, dar nu numai (vezi X Fragil de exemplu). Mulți oameni de știință spun că acest termen este incorect. Probabil că ar fi mai corect să spunem „autism de etiologie genetică de înțeles” sau ceva de genul.
Foarte recenzie buna cele mai cunoscute forme de autism cu o „etiologie genetică de înțeles” - i.e. Formele sindromice de autism pot fi găsite pe blogul lui Emily Casanova
- Partea 1: Sindroame Timothy, Smith-Lemli-Opitz, CHARGE, Cornelia de Lange, Lujan-Fryns
Încă o dată despre penetrare
Penetranța este un termen care se referă la procentul de purtători ai unei mutații care manifestă (în acest caz) autism. Revizuirea din capitolul anterior arată că penetranța pentru formele sindromice de autism este de 60 până la 90 la sută.
Penetranța de 92% pentru idic(15) este descrisă drept „uimitoare”. Da, asta e mult. Acest lucru este mai mult decât suficient pentru a considera această variantă genetică ca fiind cauza autismului – dacă se găsește la un copil și simptomele sunt consistente. Acest lucru dă, de asemenea, motive să credem că, dacă această mutație este exclusă în PGD, următorul copil aceiași părinți cu autism nu vor exista.
SNP

Acum să ajungem la cele mai mici variații genetice conținute într-o singură nucleotidă, o „litera.” Există câteva milioane de puncte în genomul uman în care variațiile dintr-o „litera” a codului genei sunt comune; Totul în jur este stabil, dar în aceste locuri specifice este diferit pentru diferiți oameni. Mai mult, din anumite motive, astfel de variații sunt aproape întotdeauna bialelice, adică există doar două variante de „litere” din patru și sunt prezente într-o proporție foarte mare a populației - adesea o variantă este în 60% și celălalt în 40%. Aceasta nu este o mutație, ci un polimorfism, dacă vă amintiți partea 7. (pentru ca un SNP să fie recunoscut oficial ca atare, prevalența sa trebuie să fie de cel puțin 1%)
Adică, acestea nu sunt „mutații”, nu erori ADN. Aceasta este norma și variațiile ei.
Astfel de variații se numesc SNP-uri (a se citi „snip”), desemnate prin numere precum rs2320030, iar aproximativ 10 milioane dintre ele au fost găsite și descrise.
Studii de asociere la nivelul genomului
O analiză care arată care variante SNP (alele) sunt prezente la o anumită persoană este efectuată pe un cip SNP. Este o placă pe care mostrele de genom uman sunt „imprimate” în jurul SNP-urilor populare, ADN-ul subiectului de testat (adesea o probă de salivă) este aplicat pe această placă și apoi se uită unde „se lipește” și unde nu 't. Populara analiză 23andme face exact asta și generează aproximativ 1 milion de astfel de SNP. Aceasta se numește „genotipizare” și este destul de ieftină, motiv pentru care acum este folosită des (și adesea inadecvat). Ieftin pentru că SNP-urile sunt bine cunoscute, iar această analiză este convenabilă de făcut on-line, în cantități mari.
Această accesibilitate a dat naștere unui tip special de cercetare, Genome-Wide Association Studies (GWAS), în care un grup de purtători ai unei trăsături moștenite și un grup de control sunt luați, toți sunt genotipați și apoi caută să vadă dacă există este orice SNP pe care primii îl au într-o variantă, iar pentru al doilea - într-un mod diferit.
Problema aici este în statistică: deoarece există milioane de SNP-uri, există o probabilitate mare să vedem o conexiune acolo unde nu există: doar de exemplu, într-un grup de 30 de persoane cu autism va exista un SNP care va apărea „ corect” de 30 de ori. Dacă aruncați o monedă de un milion de ori, aceasta va ateriza pe marginea ei :) Această problemă poate fi rezolvată folosind metode statistice și rezolvată cu succes. Cu toate acestea, în general, rezultatele GWAS pentru autism sunt prost replicate.
Ce înseamnă prezența SNP?

Efectul uneia sau celeilalte variante SNP este de obicei neglijabil. Nu poate fi altfel, întrucât toate sunt, prin definiție, foarte frecvente în populație. Cu toate acestea, există boli (de exemplu, fibroza chistică) pentru care sunt decisive (dar de obicei nu într-un singur SNP: de obicei prezența unei variante „rele” crește statistic riscul tulburării, să zicem, cu 2%).
Nici un singur SNP nu s-a găsit să fie asociat cu autismul în vreun mod semnificativ. Apropo, cele care sunt conectate cel puțin oarecum sunt, de regulă, nu în gene, ci în elementele de control ale ADN-ului - adică. nu în exom.
Prin urmare, toate testele care folosesc genotiparea SNP și calculează un „risc de autism” semnificativ sunt cel mai probabil șarlamători. Există destul de multe site-uri care oferă să descărcați rezultatele 23andme și să obțineți rețete „personalizate” pentru tratarea autismului. Cel mai faimos este „Dr.” Yasko, care promovează prescripțiile pentru „mutații” MTHFR. Escrocherie. Încă o dată, acestea nu sunt mutații. MTHFR este o genă importantă în metabolismul folaților și este probabil asociată cu autismul, dar nu este la nivelul SNP de căutat.
Pentru dreptate, trebuie spus că o combinație de SNP-uri (sute de SNP-uri diferite, fiecare dintre ele aduce o contribuție minusculă, dar aduce ceva semnificativ) pare capabilă să explice schizofrenia (spre deosebire de autism) - care este ceea ce aproape aceiași oameni încearcă să identifice Proiectul Genomului Uman și aproape în egală măsură eforturile herculeene. Dar acolo vorbim despre sute de snips.
Mutații de novo

Mutațiile „adevărate” care nu trăiesc permanent într-o populație, mutațiile care apar la indivizi specifici și sunt transmise copiilor lor (sau apar la copii) se numesc mutații de novo. Acestea pot fi suprafețe mari (CNV), mici inserții și ștergeri (indels) sau „litere” individuale. Acest post este despre ultimele două tipuri.
Pentru a le detecta, testele pe cipuri ieftine sunt nepotrivite; genomul trebuie citit succesiv sau, așa cum se numește, secvențial. Puteți citi atât întregul genom - secvențierea întregului genom (costisitor), cât și numai exomul, doar partea care codifică proteine - secvențierea exomului (relativ ieftin, 1000 USD de persoană). Prin urmare, aproape niciodată nu este utilizat în practica clinică.
În principiu, există destul de multe astfel de mutații de novo - fiecare părinte transmite cel puțin 100 dintre acestea copilului, dar doar o mică parte cade în locuri semnificative din ADN. Publicații recente și respectate (Iossifov et al 2012) estimează o contribuție cheie a mutațiilor de novo în aproximativ 10% din cazurile de autism. Dacă ați fost la conferința din mai, a existat o discuție a lui Nagwa Meguid despre „O nouă genă și o formă unică de autism” - doar un astfel de caz, mutații de novo (diverse) în gena BCKDK.
Totuși, chiar și în această poveste simplă: aici este o mutație, aici afectează o genă importantă pentru funcționarea creierului, aici am demonstrat că această genă afectează autismul - există un mare mister. Dacă, spre deosebire de Ivan Iosifov, studiem familii cu nu una, ci două persoane autiste, așa cum au făcut Yuen și colegii în acest an, atunci se dovedește că reușesc să preia mutații diferite de la aceiași părinți.
Rezumăm și clasificăm tipurile de „mutații”
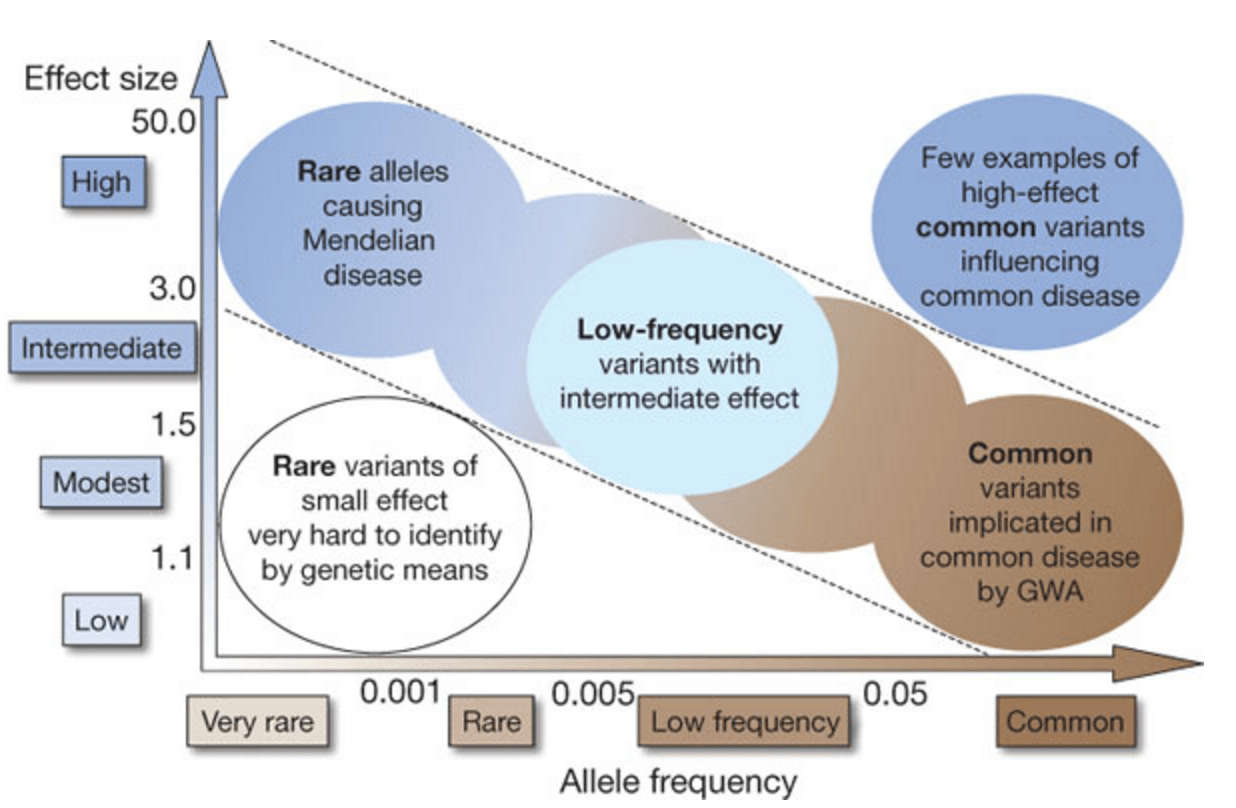
În părțile anterioare am trecut pe scurt tipuri variate variante genetice în ADN. De la dimensiuni mari, având de obicei un efect vizibil, până la foarte mici.
Și regula aici este: anomaliile mari sunt de obicei rare, dar au un efect vizibil asupra multor aspecte ale fenotipului; variantele mici sunt adesea comune în populație, dar fiecare dintre ele are un efect neglijabil.
Acest lucru poate fi rezumat într-o diagramă ca aceasta, unde linia orizontală este prevalența alelei (varianta genetică), de la rar la comun, iar linia verticală este efectul manifestat la purtători.
Trebuie remarcat faptul că pentru autism și schizofrenie, chiar și cele mai „puternice” CNV nu au efect menedelian, crescând de obicei riscul de a dezvolta o boală detectabilă clinic de la 1% la 3%-10%. Se numeste penetranta incompleta. Poți fi purtător al cunoscutului „CNV schizofrenic”, poți avea dismorfisme și anomalii ale sistemului endocrin caracteristice acestuia, dar să nu ai schizofrenie, deși poți prezenta unele anomalii subclinice.
În consecință, efectul individual al polimorfismelor de tip SNP este în general neglijabil și nu are aproape nicio semnificație practică pentru diagnosticarea autismului.
| Program educațional despre genetica autismului |
|---|
| I. Program educativ privind genetica autismului II. Genetica si gemeni III. Tipuri de mutații IV. |
Detectarea mutației denovo în gena distrofinei și semnificația acesteia pentru consilierea genetică medicală în distrofia musculară Duchenne
(observare clinica)
Muravleva E.A., Starodubova A.V., Pyshkina N.P., Duysenova O.S.
Conducător științific: Doctor în Științe Medicale conf. univ. Kolokolov O.V.
GBOU VPO Saratov State Medical University numit după. IN SI. Razumovsky Ministerul Sănătății al Federației Ruse
Catedra de Neurologie, Facultatea de Formare Pedagogică și PPS numită după. K.N. Tretiakov
Introducere. Distrofia musculară Duchenne (DMD) este una dintre cele mai frecvente boli neuromusculare moștenite. Prevalența sa este de 2-5: 100.000 de populație, frecvența populației este de 1: 3500 de băieți nou-născuți. Această formă de distrofie musculară a fost descrisă pentru prima dată de Edward Meryon (1852) și Guillaume Duchenne (1861).
Boala este caracterizată printr-un tip de moștenire recesiv legat de X și un curs sever, progresiv. DMD este cauzată de o mutație a genei distrofinei, al cărei locus este localizat pe Xp21.2. Aproximativ 30% din cazuri sunt cauzate de mutații de novo, 70% - de purtarea mutației de către mama probandului. Distrofina este responsabilă pentru conectarea citoscheletului fiecărei fibre musculare la lamina bazală subiacentă (matricea extracelulară) printr-un complex proteic care constă din multe subunități. Absența distrofinei duce la pătrunderea excesului de calciu în sarcolemă ( membrana celulara). Fibrele musculare suferă necroză, țesutul muscular este înlocuit cu țesut adipos, precum și țesut conjunctiv.
Diagnosticul modern al DMD se bazează pe evaluarea conformității manifestărilor bolii cu criteriile clinice, anamnestice și instrumentale de laborator (creatinkinaza serică (SKK), electroneuromiografie (ENMG), examenul histochimic al biopsiei musculare), analiza genealogică și genetică moleculară. date de cercetare.
Efectuarea consilierii genetice medicale în multe familii astăzi ajută la prevenirea nașterii unui copil bolnav. Diagnosticul ADN prenatal pentru primele etape sarcina în familiile cu un copil care suferă de DMD, va permite alegerea unor tactici suplimentare pentru părinți și, eventual, întreruperea timpurie a sarcinii dacă boala este prezentă la făt.
În unele cazuri, tabloul clinic este observat la femeile care sunt purtătoare heterozigote ale genei mutante sub formă de mușchi măriți ai gambei, slăbiciune musculară moderată, scăderea reflexelor tendinoase și periostale; conform studiilor paraclinice, nivelul CCS crește. În plus, manifestările clinice clasice ale DMD pot apărea la femeile cu sindrom Shereshevsky-Turner (genotip 45, XO).
Exemplu clinic. Clinica noastră vede un băiețel de 7 ani, K., care se plânge de slăbiciune a mușchilor brațelor și picioarelor, oboseală la mers îndelungat. Mama copilului constată căderi periodice, dificultăți în urcat pe scări, mers afectat (ca de rață), dificultăți de ridicare din poziție așezată și creșterea volumului mușchilor gambei.
Dezvoltarea timpurie a copilului a decurs fără caracteristici speciale. La vârsta de 3 ani, cei din jurul lui au observat tulburări ale funcțiilor motorii sub formă de dificultăți la urcarea scărilor, la ridicarea în picioare, copilul nu a luat parte la jocuri în aer liber și a început să obosească rapid. Apoi mersul de tip „rață” s-a schimbat. Dificultățile au crescut la ridicarea dintr-o poziție așezată sau din poziție culcat: „scara” pas cu pas, cu utilizarea activă a mâinilor. Treptat, a devenit vizibilă o creștere a volumului gambei și a altor câțiva mușchi.
La un examen neurologic, semnul clinic principal este tetrapareza periferică proximală simetrică, mai pronunțată la nivelul picioarelor (forța musculară în părțile proximale ale extremităților superioare - 3-4 puncte, în părțile distale - 4 puncte, în părțile proximale ale extremitățile inferioare - 2-3 puncte, în părțile distale - 4 puncte). Mersul s-a schimbat într-un tip „de rață”. Folosește tehnici auxiliare („miopatice”), de exemplu, ridicarea în picioare cu o scară. Tonusul muscular este redus, nu există contracturi. Hipotrofia mușchilor pelvini și ai centurii scapulare. Caracteristici „miopatice”, de exemplu sub forma unui spațiu interscapular larg. Există pseudohipertrofie a mușchilor gambei. Reflexe tendinoase și periostale - fără diferențe semnificative între părți; bicipital - scăzut, tricipital și carporradial - vioitate medie, genunchi și Ahile - scăzut. Pe baza datelor clinice, a fost suspectată DMD.
La studierea CCS, nivelul acestuia a fost de 5379 unități/l, ceea ce este de 31 de ori mai mare decât norma (norma este de până la 171 unități/l). Potrivit ENMG, au fost înregistrate semne care sunt mai caracteristice unui proces muscular primar moderat în curs. Astfel, datele obținute au confirmat prezența DMD la pacient.
Pe lângă proband, au fost examinați părinții și sora mai mare. Niciuna dintre rudele probandului nu a avut manifestări clinice de DMD. Cu toate acestea, mama a observat o ușoară creștere în volum a mușchilor gambei. Conform analizei genealogice, probanda este singura persoană bolnavă din familie. Nu se poate exclude faptul că mama copilului și sora probandului sunt purtători heterozigoți ai genei mutante (Fig. 1).
Orez. 1 Pedigree
Ca parte a consilierii genetice medicale, familia lui K. a fost examinată pentru prezența/absența delețiilor și dublărilor în gena distrofinei. Analiza genetică moleculară în laboratorul de diagnosticare ADN al Centrului de Cercetare de Stat din Moscova al Academiei Ruse de Științe Medicale a relevat o ștergere a exonului 45 în probandul K., care confirmă în cele din urmă diagnosticul clinic stabilit de DMD. Mama nu a avut deleția exonului 45 identificată la fiul ei. În urma analizei, ștergerea surorii a exonului 45, identificat la fratele ei, nu a fost găsită. Prin urmare, la persoana studiată, mutația are cel mai probabil o origine de novo, dar poate fi și rezultatul mozaicismului germinativ la mamă. În consecință, cu o mutație de novo, riscul de a avea un copil bolnav la o mamă va fi determinat de frecvența populației acestei mutații (1:3500, <1%), care este semnificativ mai mică decât în cazul unui tip recesiv legat de X. de moştenire (50% din băieţi). Deoarece este imposibil de exclus complet faptul că mutația poate fi rezultatul mozaicismului germinal, în care moștenirea conform legilor mendeliane este încălcată, se recomandă diagnosticul prenatal în timpul sarcinii ulterioare la mama și sora probanului.
Concluzie.În prezent, medicul are un arsenal larg de medicamente simptomatice utilizate în tratamentul DMD, cu toate acestea, în ciuda progreselor științei, tratamentul etiologic al DMD nu a fost încă dezvoltat, medicamente eficiente Nu există tratament de înlocuire pentru DMD. Conform cercetărilor recente cu celule stem, există vectori promițători care pot înlocui țesutul muscular deteriorat. Cu toate acestea, în prezent, este posibil doar tratamentul simptomatic care vizează îmbunătățirea calității vieții pacientului. În această privință diagnostic precoce DMD joacă un rol vital pentru consilierea medicală genetică în timp util și pentru alegerea unor tactici ulterioare de planificare familială. Pentru diagnosticul ADN prenatal, prelevarea de vilozități coriale (CVS) poate fi efectuată la 11-14 săptămâni de sarcină, amniocenteza poate fi folosită după 15 săptămâni, iar prelevarea de sânge fetal se poate face în jurul a 18 săptămâni. Dacă testarea este efectuată la începutul sarcinii, întreruperea timpurie a sarcinii este posibilă dacă fătul are boala. În unele cazuri, este recomandabil să se efectueze diagnostice ADN preimplantare urmate de fertilizarea in vitro.
Concluzii. Pentru a asigura depistarea precoce și prevenirea DMD, este necesar să se utilizeze mai pe scară largă metodele de diagnostic genetic molecular; spori vigilența medicilor practicanți cu privire la această patologie. Cu o mutație de novo, riscul mamei de a avea un copil bolnav este determinat de frecvența populației a mutației genei distrofinei. În cazurile în care mama probandului poartă mutația, diagnosticul ADN prenatal sau perimplantare este necesar în scopul planificării familiale.
Citogenetica medicală este studiul cariotipului uman în condiții normale și patologice. Această direcție a apărut în 1956, când Tio și Levan au îmbunătățit metoda de preparare a preparatelor cromozomilor metafazici și au stabilit pentru prima dată numărul modal de cromozomi (2n=46) într-un set diploid. În 1959, a fost descifrată etiologia cromozomială a unui număr de boli - sindromul Down, sindromul Klinefelter, sindromul Shereshevsky-Turner și alte câteva sindroame trisomale autosomale. Dezvoltarea ulterioară a citogeneticii medicale la sfârșitul anilor 1960 s-a datorat apariției metodelor de colorare diferențială a cromozomilor metafazici, care au făcut posibilă identificarea cromozomilor și a regiunilor lor individuale. Metodele de colorare diferențială nu au asigurat întotdeauna identificarea corectă a punctelor de întrerupere ca urmare a rearanjamentelor structurale ale cromozomilor. În 1976, Younis a dezvoltat noi metode pentru studierea lor în stadiul de prometafază, care au fost numite „metode de înaltă rezoluție”.
Utilizarea unor astfel de metode a făcut posibilă obținerea de cromozomi cu un număr diferit de segmente (de la 550 la 850) și a făcut posibilă identificarea tulburărilor care implică secțiuni mici ale acestora (microrearanjamente). De la începutul anilor 1980. citogenetica umană a intrat în noua etapa dezvoltare: analiza cromozomiala a metodelor citogenetice moleculare, hibridizarea fluorescenta in situ (FISH - Fluorescence In Situ Hybridization) a fost introdusa in practica. Această metodă este utilizată pe scară largă pentru a detecta anomalii structurale mai subtile ale cromozomilor care nu se pot distinge prin colorarea diferențială. În prezent, utilizarea diferitelor metode de analiză cromozomială face posibilă efectuarea cu succes a diagnosticului pre și postnatal al bolilor cromozomiale.
Bolile cromozomiale sunt un grup mare de afecțiuni clinic diverse, caracterizate prin multiple malformații congenitale, a căror etiologie este asociată cu modificări cantitative sau structurale ale cariotipului.
În prezent, se disting aproape 1000 de anomalii cromozomiale, dintre care peste 100 de forme au un tablou definit clinic și se numesc sindroame; contribuția lor la avorturile spontane, mortalitatea și morbiditatea neonatale este semnificativă. Prevalența anomaliilor cromozomiale în rândul avorturilor spontane este în medie de 50%, la nou-născuții cu malformații congenitale multiple severe - 33%, decesele născuți morți și perinatale cu malformații congenitale - 29%, prematurii cu malformații congenitale - 17%, nou-născuții cu malformații congenitale - 10% , decese născuți morți și perinatale - 7%, prematuri - 2,5%, toți nou-născuții - 0,7%.
Majoritatea bolilor cromozomiale sunt sporadice, apar din nou ca urmare a unei mutații genomice (cromozomiale) în gametul unui părinte sănătos sau în primele diviziuni ale zigotului și nu sunt moștenite de-a lungul generațiilor, ceea ce este asociat cu mortalitatea ridicată a pacienților din perioada pre-reproductivă. Baza fenotipică a bolilor cromozomiale sunt tulburările dezvoltării embrionare timpurii. De aceea, modificările patologice se dezvoltă chiar și în perioada prenatală de dezvoltare a organismului și fie provoacă moartea embrionului sau a fătului, fie creează principalul tablou clinic boli deja la nou-născut (cu excepția anomaliilor dezvoltării sexuale, care se dezvoltă în principal în timpul pubertății). Leziunile precoce și multiple ale sistemelor corpului sunt caracteristice tuturor formelor de boli cromozomiale. Acestea sunt dismorfia cranio-facială, malformațiile congenitale organe interneși părți ale corpului, creștere și dezvoltare lentă intrauterină și postnatală, întârziere mentală, defecte ale sistemului nervos central, cardiovascular, respirator, genito-urinar, digestiv și endocrin, precum și abateri ale stării hormonale, biochimice și imunologice. Fiecare sindrom cromozomial se caracterizează printr-un complex de malformații congenitale și anomalii de dezvoltare, care sunt inerente într-o oarecare măsură doar acestui tip de patologie cromozomială. Polimorfismul clinic al fiecărei boli cromozomiale în forma sa generală este determinat de genotipul organismului și de condițiile de mediu. Variațiile în manifestările patologiei pot fi foarte largi - de la un efect letal la abateri minore de dezvoltare. În ciuda studiului bun al manifestărilor clinice și citogeneticii bolilor cromozomiale, patogenia acestora, chiar și în termeni generali, nu este încă clară. Nedezvoltat schema generala dezvoltarea unor procese patologice complexe cauzate de anomalii cromozomiale și care conduc la apariția fenotipurilor complexe ale bolilor cromozomiale.
Principalele tipuri de anomalii cromozomiale
Toate bolile cromozomiale în funcție de tipul de mutații pot fi împărțite în două mari grupe: cele cauzate de modificări ale numărului de cromozomi cu menținerea structurii acestora din urmă (mutații genomice) și cele cauzate de modificări ale structurii cromozomilor (cromozomi). mutații). Mutațiile genomice apar din cauza nedisjuncției sau pierderii cromozomilor în timpul gametogenezei sau în stadiile incipiente ale embriogenezei. Doar trei tipuri de mutații genomice au fost găsite la om: tetraploidie, triploidie și aneuploidie. Incidența mutațiilor triploide (Zn=69) și tetraploide (4n=92) este foarte scăzută, ele se găsesc în principal printre embrionii sau fetușii avortați spontan și la născuții morți. Speranța de viață a nou-născuților cu astfel de tulburări este de câteva zile. Mutațiile genomice pe cromozomi individuali sunt numeroase; ele alcătuiesc cea mai mare parte a bolilor cromozomiale. Mai mult decât atât, dintre toate variantele de aneuploidie se găsesc doar trisomia pe autozomi, polisomia pe cromozomii sexuali (tri-, tetra- și pentasomia), iar dintre monosomii se găsește doar monosomia X.
Trisomiile sau monosomiile complete sunt mai greu de tolerat de către organism decât cele parțiale; dezechilibrele în cromozomii mari apar la născuții vii mult mai puțin frecvent decât la cei mici. Formele complete de anomalii cromozomiale provoacă anomalii semnificativ mai grave decât cele din mozaic. Monosomiile autozomale sunt foarte rare în rândul născuților vii; sunt forme mozaic cu o mare proporție de celule normale. Faptul valorii genetice relativ scăzute a regiunilor heterocromatice ale cromozomilor a fost dovedit. De aceea se observă trisomii complete la născuții vii în acei autozomi care sunt bogați în heterocromatină - 8, 9, 13, 14, 18, 21, 22 și X. Aceasta explică toleranța bună de către pacienți chiar și la o doză triplă de Y- materialul cromozomic și pierderea aproape completă a umărului său lung Monosomia completă pe cromozomul X, compatibilă cu viața postnatală, care duce la dezvoltarea sindromului Shereshevsky-Turner, precum și tetra- și pentasomia, sunt observate numai pe cromozomul X, care este heterocromatic.
Mutațiile cromozomiale sau rearanjamentele cromozomiale structurale sunt tulburări de cariotip, însoțite sau nu de un dezechilibru al materialului genetic în cadrul unuia sau mai multor cromozomi (rearanjamente intra și intercromozomiale).
În majoritatea covârșitoare a cazurilor, mutațiile cromozomiale structurale sunt transmise descendenților de către unul dintre părinți, al cărui cariotip conține o rearanjare cromozomială echilibrată. Acestea includ translocarea reciprocă (reciprocă) echilibrată fără pierderea secțiunilor cromozomilor implicați în aceasta. Ea, ca și inversarea, nu provoacă fenomene patologice la purtător. Cu toate acestea, în timpul formării gameților din purtători de translocații și inversiuni echilibrate, se pot forma gameți dezechilibrati. Translocarea robertsoniană - o translocare între doi cromozomi acrocentrici cu pierderea brațelor lor scurte - duce la formarea unui cromozom metacentric în loc de doi cromozomi acrocentrici. Purtătorii acestei translocații sunt sănătoși deoarece pierderea brațelor scurte a doi cromozomi acrocentrici este compensată de activitatea acelorași gene în restul de 8 cromozomi acrocentrici. În timpul maturării celulelor germinale distribuție aleatorie(în timpul diviziunii celulare) a doi cromozomi rearanjați și omologii acestora duce la apariția mai multor tipuri de gameți, dintre care unii sunt normali, alții conțin o astfel de combinație de cromozomi care, la fecundare, produce un zigot cu un cariotip rearanjat echilibrat, în timp ce altele produc zigoti dezechilibrati cromozomial in timpul fertilizarii.
Cu un set de cromozomi dezechilibrat (deleții, duplicări, inserții), fătul dezvoltă patologii clinice severe, de obicei sub forma unui complex de malformații congenitale. Lipsa materialului genetic provoacă defecte de dezvoltare mai grave decât un exces al acestuia.
Mult mai rar, aberațiile structurale apar de novo. Părinții unui pacient cu o tulburare cromozomială sunt de obicei normali cariotipic. Boala cromozomală în aceste cazuri apare de novo ca urmare a transmiterii de la unul dintre părinți a unei mutații genomice sau cromozomiale care apare o dată în unul dintre gameți, sau o astfel de mutație apare deja în zigot. Acest lucru nu exclude reapariția unei tulburări cromozomiale la copiii dintr-o anumită familie. Există familii predispuse la cazuri repetate de nondisjuncție cromozomială. Mutațiile care au apărut de novo reprezintă aproape toate cazurile de trisomii și monosomii complete cunoscute. Principalul mecanism pentru apariția rearanjamentelor structurale de orice tip este o rupere a unuia sau mai multor cromozomi cu reunificarea ulterioară a fragmentelor rezultate.
Indicații clinice pentru diagnosticul citogenetic
Metoda de cercetare citogenetică ocupă un loc de frunte între metodele de diagnostic de laborator în consilierea genetică medicală și diagnosticul prenatal. Cu toate acestea, ar trebui să respectați cu strictețe obiectivul
indicaţii pentru trimiterea pacienţilor pentru testarea cariotipului.
Principalele indicații pentru diagnosticul prenatal:
anomalie cromozomiala la copilul anterior din familie;
copil născut mort cu o anomalie cromozomială;
rearanjamente cromozomiale, mozaicism cromozomial sau aneuploidie pe cromozomii sexuali la părinți;
rezultatele unui test de ser sanguin matern care indică un risc crescut de anomalie cromozomială la făt (grup de risc);
vârsta mamei;
anomalii fetale detectate prin examinare cu ultrasunete;
suspiciunea de mozaicism la făt în timpul unui studiu citogenetic anterior;
sindrom de instabilitate cromozomiala suspectat.
Testarea cariotipului pentru diagnosticul postnatal este recomandată dacă pacienta are:
amenoree primară sau secundară sau menopauză precoce;
spermogramă anormală - azoospermie sau oligospermie severă;
anomalii de creștere semnificative clinic (scăzut, crestere mare) și dimensiunea capului (micro-, macrocefalie);
organe genitale anormale;
fenotip anormal sau dismorfie;
malformații congenitale;
retard mintal sau tulburări de dezvoltare;
manifestări ale sindromului de deleție/microdeleție/duplicare;
Boală recesivă legată de X la femei;
manifestări clinice ale sindroamelor de instabilitate cromozomială;
la monitorizarea după transplant de măduvă osoasă.
Studiile citogenetice ar trebui efectuate într-un cuplu căsătorit:
cu anomalii cromozomiale sau opțiuni neobișnuite cromozomi la făt detectați în timpul diagnosticului prenatal;
avorturi repetate (3 sau mai multe); naștere mortină, moarte fetală neonatală, incapacitatea de a examina fătul afectat;
copilul are o anomalie cromozomială sau o variantă cromozomială neobișnuită;
infertilitate cu etiologie necunoscută.
Indicația pentru cercetarea citogenetică este prezența rudelor pacientului:
rearanjamente cromozomiale;
retard mintal probabil de origine cromozomială;
pierderi de reproducere, malformații congenitale ale fătului sau naștere morta de origine necunoscută.
Indicații pentru cercetarea folosind metoda FISH:
suspiciunea de sindrom de microdeleție, pentru care sunt disponibile diagnostice citogenetice moleculare (disponibilitatea sondelor ADN adecvate);
risc crescut de sindrom de microdeleție pe baza datelor anamnestice;
semne clinice care sugerează mozaicismul datorat unui anumit sindrom cromozomial;
afecțiuni după transplantul de măduvă osoasă, când donatorul și primitorul sunt de sexe diferite;
suspiciunea unei anomalii cromozomiale în timpul unui studiu citogenetic standard, când metoda FISH poate fi utilă pentru continuarea
clarificarea naturii anomaliei, sau în situațiile în care există manifestări clinice caracteristice;
prezența unui cromozom marker supranumerar;
suspiciunea de rearanjare cromozomială ascunsă.
Metoda FISH pentru analiza metafazelor este indicată:
cu cromozomi marker;
material suplimentar de origine necunoscută pe cromozom;
rearanjamente cromozomiale;
pierderea suspectată a unui segment cromozomial;
mozaicism.
Metoda FISH pentru analiza nucleelor de interfaza este indicata:
cu anomalii cromozomiale numerice;
duplicari;
diviziuni;
rearanjamente cromozomiale;
determinarea sexului cromozomial;
amplificarea genelor.
Metode de cercetare citogenetică:
Cercetare și descriere trasaturi caracteristice cromozomii metafazici sunt deosebit de importanți pentru citogenetica practică. Cromozomii individuali dintr-un grup sunt recunoscuți folosind tehnici de colorare diferențială. Aceste metode fac posibilă detectarea eterogenității structurii cromozomilor de-a lungul lungimii, determinată de caracteristicile complexului principalelor componente moleculare ale cromozomilor - ADN și proteine. Problema recunoașterii cromozomilor individuali într-un cariotip este importantă pentru dezvoltarea diagnosticului citogenetic al bolilor cromozomiale la om.
Metodele de cercetare citogenetică sunt împărțite în directe și indirecte. Metodele directe sunt utilizate în cazurile în care este nevoie de un rezultat rapid și este posibil să se obțină preparate de cromozomi ai celulelor care se divid în organism. Metodele indirecte includ, ca pas obligatoriu, cultivarea mai mult sau mai puțin pe termen lung a celulelor în artificial medii nutritive. Metodele care includ cultivarea pe termen scurt (de la câteva ore la 2-3 zile) ocupă o poziție intermediară.
Obiectul principal al cercetării citogenetice prin metode directe și indirecte este stadiul metafazic al mitozei și diferitele etape ale meiozei. Metafaza mitozei este subiectul principal al cercetării citogenetice, deoarece în acest stadiu este posibilă identificarea precisă a cromozomilor și detectarea anomaliilor acestora. Cromozomii din meioză sunt examinați pentru a detecta anumite tipuri de rearanjamente care, prin natura lor, nu sunt detectate în metafaza mitozei.
Material biologic pentru studii citogenetice. Prelucrarea culturilor celulare. Prepararea preparatelor cromozomiale
Celulele din orice țesut disponibil pentru biopsie pot fi folosite ca material pentru obținerea cromozomilor umani și studierea acestora. Cele mai frecvent utilizate sunt sângele periferic, fibroblastele pielii, măduva osoasă, celulele lichidului amniotic și celulele viloase coriale. Limfocitele din sângele periferic uman sunt cele mai accesibile pentru cercetarea cromozomilor.
În prezent, aproape toate laboratoarele din lume folosesc o metodă care utilizează sânge integral periferic pentru a cultiva limfocite. Sângele în cantitate de 1-2 ml este prelevat în prealabil din vena cubitală într-un tub steril sau o sticlă cu soluție de heparină. Sângele într-un flacon poate fi păstrat timp de 24-48 de ore la frigider la o temperatură de 4-6 °C. Cultura limfocitelor se efectuează într-o cameră specială sau în camera de lucru sub o hota cu flux laminar in conditii sterile. Astfel de condiții sunt obligatorii pentru a preveni introducerea florei patogene în hemocultură. Dacă există suspiciunea de contaminare a sângelui sau a altor materiale, este necesar să se adauge antibiotice la amestecul de cultură. Flacoanele cu amestecul de cultură sunt incubate într-un termostat la o temperatură de +37 °C timp de 72 de ore (creșterea și diviziunea celulară activă sunt în curs). Scopul principal al tehnicilor metodologice atunci când se prelucrează culturile celulare și se prepară preparatele cromozomiale din acestea este de a obține pe preparat un număr suficient de plăci metafazice cu o astfel de răspândire a cromozomilor încât să fie posibilă estimarea lungimii, formei și a altor caracteristici morfologice ale fiecăruia. cromozom din set.
Acumularea de celule în metafaza mitozei și producerea de plăci de înaltă calitate pe preparat are loc utilizând o serie de proceduri secvențiale:
colchinizare - expunerea celulelor la citostatice colchicină sau colcemid, blocând mitoza în stadiul de metafază;
hipotonizarea culturilor;
fixarea celulelor cu un amestec de alcool metilic și acid acetic;
aplicarea unei suspensii celulare pe o lamă de sticlă.
Colchinizarea culturilor celulare se realizează cu 1,5-2 ore înainte de începerea fixării. După administrarea colchicinei, sticlele de cultură celulară continuă să incubeze în termostat. La sfârșitul incubației, amestecul de cultură din fiecare sticlă este turnat în tuburi de centrifugă curate și supus centrifugării. Apoi, la sedimentul celular se adaugă o soluție hipotonică de clorură de potasiu, preîncălzită la o temperatură de +37 °C.
Hipotonizarea se efectuează într-un termostat la o temperatură de +37 °C timp de 15 minute. O soluție hipotonică KCI promovează o mai bună răspândire a cromozomilor pe o lamă de sticlă. După hipotonizare, celulele sunt sedimentate prin centrifugare și supuse fixării. Fixarea se realizează cu un amestec de alcool metilic (sau etilic) și acid acetic.
Etapa finală este pregătirea preparatelor cromozomiale pentru a obține plăci metafazice bine răspândite, menținând în același timp integritatea și completitudinea setului de cromozomi din fiecare dintre ele. O suspensie celulară este aplicată pe lamele umede, răcite, după care lamelele sunt uscate la temperatura camerei și etichetate.
Metode de colorare diferențială a cromozomilor
Din 1971, în citogenetică s-au răspândit metode care fac posibilă colorarea diferențială a fiecărui cromozom dintr-un set în funcție de lungimea acestuia. Semnificația practică a acestor metode este că colorarea diferențială permite identificarea tuturor cromozomilor umani datorită modelului de colorare longitudinal specific pentru fiecare cromozom. Orice vopsea constând dintr-un colorant de bază poate fi potrivită pentru colorare, deoarece principalul substrat colorant al cromozomilor este complexul ADN-proteină. În practica cercetării citogenetice cea mai mare aplicație a primit următoarele metode.
Metoda de colorare cu G este cea mai comună metodă datorită simplității, fiabilității și disponibilității reactivilor necesari. După colorare, fiecare pereche de cromozomi capătă striații în lungime datorită alternanței segmentelor heterocromatice (întunecate) și eucromatice (luminoase) colorate diferit, care sunt denumite de obicei segmente G. Metoda de colorare cu C oferă identificarea doar a anumitor regiuni ale cromozomilor. Acestea sunt regiuni ale heterocromatinei localizate în regiunile pericentromerice ale brațelor lungi ale cromozomilor 1, 9 și 16 și în brațul lung al cromozomului Y, precum și în brațele scurte ale cromozomilor acrocentrici. Metoda R a preparatelor de cromozomi de colorare arată o imagine a segmentării diferenţiale inversă cu metoda G. Această metodă colorează bine segmentele distale ale cromozomilor, ceea ce este foarte important atunci când se identifică mici rearanjamente care implică secțiunile terminale. Metoda de colorare Q oferă colorare fluorescentă diferențială a cromozomilor individuali ai setului, vă permite să identificați fiecare pereche de omologi și, de asemenea, să determinați prezența unui cromozom Y în nucleele de interfază prin strălucirea corpului cromatinei Y.
Principiile analizei cromozomilor
O etapă obligatorie a studiului este o analiză vizuală a cromozomilor la microscop folosind o mărire de o mie de ori (x1000) cu oculare x10 și o lentilă de imersie x100. Evaluarea calității și adecvarea preparatelor cromozomiale pentru cercetare, precum și selecția plăcilor metafazate pentru analiză, se efectuează la o mărire mică (x100). Pentru studiu sunt selectate plăci metafază bine colorate, complete, cu o bună răspândire a cromozomilor. Cercetătorul numără numărul total de cromozomi și evaluează structura fiecărui cromozom comparând striațiile omologilor, precum și comparând modelul observat cu hărțile (schemele) citogenetice ale cromozomilor.
Utilizarea sistemelor computerizate de analiză a imaginilor simplifică foarte mult sarcina unui citogenetician, îmbunătățește calitatea muncii sale și oferă posibilitatea de a documenta rapid și ușor rezultatele cercetării. Pentru a asigura o calitate înaltă a muncii, se recomandă ca doi specialiști să participe la studiul citogenetic al fiecărei probe. Documentul care confirmă studiul este protocolul, care indică coordonatele celulelor examinate, numărul de cromozomi din fiecare dintre ele, rearanjamentele detectate, formula și concluzia cariotipului, precum și numele de familie al pacientului, data și numărul studiu, numele și semnătura medicului (medicilor) care au efectuat studiul . Diapozitivele și imaginile cromozomiale ar trebui să fie salvate pentru revizuire ulterioară.
REGULI DE BAZĂ PENTRU DESCRIEREA ANOMALIILOR CROMOZOMIALE CONFORM SISTEMULUI INTERNAȚIONAL DE NOMENCLATURĂ CITOGENETICĂ
Înregistrarea formulei cariotipului trebuie efectuată în conformitate cu versiunea actuală a Sistemului internațional pentru nomenclatura citogenetică umană. Mai jos luăm în considerare aspectele utilizării nomenclaturii care sunt cel mai des întâlnite în practica citogenetică clinică.
Numărul și morfologia cromozomilor:
Într-un cariotip, cromozomii sunt împărțiți în șapte grupuri ușor de distins (A-G) în funcție de dimensiunea și poziția centromerului. Autozomii sunt cromozomii de la 1 la 22, cromozomii sexuali sunt X și Y.
Grupa A (1-3) - cromozomi metacentrici mari care se pot distinge unul de celălalt prin dimensiune și poziția centromerului.
Grupa B (4-5) - cromozomi mari submetacentrici.
Grupa C (6-12, X) - cromozomi metacentrici și submetacentrici de mărime medie. Cromozomul X este unul dintre cei mai mari cromozomi din acest grup.
Grupa D (13-15) - cromozomi acrocentrici de dimensiuni medii cu sateliți.
Grupa E (16-18) - cromozomi metacentrici și submetacentrici relativ mici.
Grupa F (19-20) - cromozomi metacentrici mici.
Grupa G (21-22, Y) - cromozomi mici acrocentrici cu sateliți. Cromozomul Y nu are sateliți.
Fiecare cromozom constă dintr-o serie continuă de dungi, care sunt situate de-a lungul lungimii brațelor cromozomilor în zone (secțiuni) strict limitate. Regiunile cromozomiale sunt specifice fiecărui cromozom și sunt esențiale pentru identificarea lor. Benzile și regiunile sunt numerotate în direcția de la centromer la telomer pe lungimea fiecărui braț. Regiunile sunt secțiuni ale unui cromozom situat între două benzi adiacente. Pentru a desemna brațele scurte și lungi ale cromozomilor se folosesc următoarele simboluri: p - braț scurt și q - braț lung. Centromerul (sep) este desemnat prin simbolul 10, partea centromerului adiacentă brațului scurt este p10, iar brațului lung este q10. Regiunea cea mai apropiată de centromer este desemnată cu numărul 1, regiunea următoare cu numărul 2 etc.
Simbolismul din patru cifre este folosit pentru a desemna cromozomi:
primul caracter - numărul de cromozomi;
Al 2-lea caracter (p sau q) - brațul cromozomului;
Al 3-lea caracter - numărul raionului (secțiunii);
Al 4-lea caracter este numărul benzii din această zonă.
De exemplu, intrarea 1p31 indică cromozomul 1, brațul său scurt, regiunea 3, banda 1. Dacă banda este împărțită în sub-benzi, un punct este plasat după desemnarea benzii, atunci se scrie numărul fiecărei sub-benzi. Sub-benzile, ca dungile, sunt numerotate în direcția de la centromer la telomer. De exemplu, în banda 1p31 există trei sub-benzi: 1p31.1, 1p31.2 și 1p31.3, dintre care subbanda 1p31.1 este proximală de centromer, iar subbanda 1p31.3 este distală. Dacă sub-benzile sunt mai departe subdivizate în părți, acestea sunt numerotate cu numere fără punctuație. De exemplu, subbanda 1р31.1 este împărțită în 1р31.11, 1р31.12 etc.
PRINCIPII GENERALE PENTRU DESCRIEREA CARIOTIPULUI NORMAL ȘI ANORMAL
În descrierea cariotipului, primul punct indică numărul total de cromozomi, inclusiv cromozomi sexuali. Primul număr este separat de restul intrării printr-o virgulă, apoi se notează cromozomii sexuali. Autozomii sunt desemnați numai în cazuri de anomalii.
Un cariotip uman normal arată astfel:
46,XX - cariotip normal al unei femei;
46,XY este cariotipul normal al unui bărbat.
În cazul anomaliilor cromozomiale se înregistrează mai întâi anomaliile cromozomiale sexuale, apoi anomaliile autozomale în ordine crescătoare a numerelor și indiferent de tipul anomaliei. Fiecare anomalie este separată prin virgulă. Denumirile de litere sunt folosite pentru a descrie cromozomii rearanjați structural. Cromozomul implicat în rearanjare se scrie între paranteze după simbolul care indică tipul de rearanjare, de exemplu: inv(2), del(4), r(18). Dacă în rearanjare sunt implicați doi sau mai mulți cromozomi, între numărul fiecărui cromozom este plasat un punct și virgulă (;).
Semnele (+) sau (-) sunt plasate în fața unui cromozom pentru a indica o anomalie, indicând un cromozom suplimentar sau lipsă (normal sau anormal), de exemplu: +21,-7,+der(2). Ele sunt, de asemenea, folosite pentru a indica o scădere sau o creștere a lungimii unui braț de cromozom după simbol (p sau q); în acest scop, semnele de mai sus pot fi folosite doar în text, dar nu și în descrierea cariotipului, de exemplu: 4p+, 5q-. Când descriem dimensiunile segmentelor heterocromatice, ale sateliților și ale filamentelor de satelit, semnul (+) (creștere) sau (-) (scădere) este plasat imediat după desemnarea simbolului corespunzător, de exemplu: 16qh+, 21ps+, 22pstk+. Semnul de multiplicare (x) este folosit pentru a descrie mai multe copii ale cromozomilor rearanjați, dar nu poate fi folosit pentru a descrie mai multe copii ale cromozomilor normali, de exemplu: 46,XX,del(6)(q13q23)x2. Pentru a indica interpretări alternative ale anomaliilor, utilizați simbolul (sau), de exemplu: 46,XX,del(8)(q21.1) sau i(8)(p10).
Cariotipurile diferitelor clone sunt separate printr-o bară oblică (/). Parantezele pătrate sunt plasate după descrierea cariotipului pentru a indica numărul absolut de celule dintr-o clonă dată. Pentru a indica motivul apariției diferitelor clone, se folosesc simbolurile mos (mozaicism - linii celulare provenite din același zigot) și chi (himeră - linii celulare provenite din diferiți zigoți), care sunt date înainte de descrierea cariotip. La listarea cariotipurilor, clona diploidă normală este întotdeauna listată ultima, de exemplu: mos47,XY,+21/46,XY; mos47,XXY/46,XY.
Dacă există mai multe clone anormale, înregistrarea se realizează în ordinea mărimii crescătoare: prima este cea mai frecvent întâlnită, apoi descrescătoare. Ultima este clona normală, de exemplu: mos45,X/47,XXX/46,XX. O notație similară este utilizată într-un cariotip care are două clone normale, de exemplu: chi46,XX/46,XY. Dacă în cariotip sunt prezente două clone anormale, dintre care una are o anomalie numerică, iar cealaltă are o rearanjare structurală, atunci clona cu anomalia numerică este înregistrată mai întâi. De exemplu: 45,X/46,X,i(X)(q10).
Când ambele clone au anomalii numerice, se înregistrează prima clona cu autozomul cu numărul de serie mai mic, de exemplu: 47,XX,+8/47,XX,+21; clona cu anomalii ale cromozomilor sexuali este întotdeauna plasată pe primul loc, de exemplu: 47,ХХХ/47,ХХ,+21.
Faptul că cariotipul este haploid sau poliploid va fi evident din numărul de cromozomi și denumiri ulterioare, de exemplu: 69,XXY. Toți cromozomii modificați trebuie desemnați în raport cu nivelul adecvat de ploidie, de exemplu: 70,XXY,+21.
Originea maternă sau paternă a unui cromozom anormal este indicată prin simbolurile mat și, respectiv, pat, după anomalia descrisă, de exemplu: 46,XX,t(5;6)(q34;q23)mat,inv(14)( q12q31)pat; 46,XX,t(5;6)(q34;q23)mat,inv(14) (q12q31)mat. Dacă se știe că cromozomii părinților sunt normali în comparație cu o anumită anomalie, aceasta este considerată una nouă și este desemnată prin simbolul denovo (dn), de exemplu: 46,XY,t(5;6)(q34). ;q23)mat,inv (14)( q12q31)dn.
Descrierea anomaliilor cromozomiale numerice:
Semnul (+) sau (-) este folosit pentru a indica pierderea sau achiziționarea unui cromozom suplimentar atunci când descriem anomalii numerice.
47,XX,+21 - cariotip cu trisomie 21.
48,XX,+13,+21 - cariotip cu trisomie 13 și trisomie 21.
45,XX,-22 - cariotip cu monosomie 22.
46,XX,+8,-21 - cariotip cu trisomie 8 și monosomie 21.
O excepție de la această regulă sunt anomaliile constituționale ale cromozomilor sexuali, care sunt scrise fără a folosi semnele (+) și (-).
45,X - cariotip cu un cromozom X (sindromul Shereshevsky-Turner).
47,XXY - cariotip cu doi cromozomi X și un cromozom Y (sindromul Klinefelter).
47,XXX - cariotip cu trei cromozomi X.
47,XYY - cariotip cu un cromozom X și doi cromozomi Y.
48,XXXY este un cariotip cu trei cromozomi X și un cromozom Y.
Descrierea anomaliilor structurale ale cromozomilor
În descrierea modificărilor structurale sunt utilizate atât sisteme de înregistrare succinte, cât și detaliate. Când se utilizează sistemul scurt, sunt indicate doar tipul de rearanjare cromozomială și punctele de întrerupere. Notați tipul de anomalie cromozomială, cromozomul implicat în anomalie și punctele de întrerupere între paranteze. Sistemul scurt nu permite o descriere fără ambiguitate a rearanjamentelor cromozomiale complexe, care sunt uneori detectate la analiza cariotipurilor tumorale.
Sistem scurt de desemnare a ajustărilor structurale
Dacă ambele brațe sunt implicate într-o rearanjare rezultată din două rupturi care apar într-un cromozom, punctul de rupere în brațul scurt este înregistrat înainte de punctul de întrerupere în brațul lung: 46,XX,inv(2)(p21q31). Când două puncte de întrerupere sunt pe același braț al cromozomului, punctul de întrerupere proximal de centromer este indicat mai întâi: 46,XX,inv(2)(p13p23). În cazul în care în rearanjare sunt implicați doi cromozomi, se indică mai întâi fie cromozomul cu un număr de serie mai mic, fie cromozomul sexual: 46,XY,t(12;16)(q13;p11.1); 46,X,t(X;18) (p11.11;q11.11).
Excepția de la regulă este rearanjarea cu trei puncte de întrerupere, când un fragment dintr-un cromozom este inserat într-o regiune a altui cromozom. În acest caz, se scrie primul cromozom receptor, iar ultimul cromozom donor, chiar dacă este un cromozom sexual sau un cromozom cu un număr de serie mai mic: 46,X,ins(5;X)(p14;q21q25); 46,XY,ins(5;2)(p14;q22q32). Dacă rearanjarea afectează un cromozom, punctele de întrerupere din segmentul în care s-a format inserția sunt indicate mai întâi. În cazul inserției directe, se înregistrează mai întâi punctul de rupere al fragmentului inserat proximal de centromer, iar apoi punctul de rupere distal. Cu o inserție inversată, este adevărat opusul.
Pentru a indica translocațiile în care sunt implicați trei cromozomi diferiți, este indicat mai întâi cromozomul sexual sau cromozomul cu un număr de serie mai mic, apoi cromozomul care a primit un fragment din primul cromozom și, în final, cromozomul care a donat fragmentul primul cromozom. 46,XX,t(9;22;17) (q34;q11.2;q22) - un fragment al cromozomului 9, corespunzător regiunii distale 9q34, transferat la cromozomul 22, la segmentul 22q11.2, un fragment de cromozom 22, corespunzătoare regiunii distale 22q11 .2 este transferat la cromozomul 17, în segmentul 17q22, iar fragmentul cromozomului 17, corespunzător regiunii distale a 17q22, este transferat la cromozomul 9, în segmentul 9q34.
Sistem detaliat pentru desemnarea modificărilor structurale. În conformitate cu un sistem de notare detaliat, rearanjamentele structurale ale cromozomilor sunt determinate de compoziția benzilor din acestea. Toate denumirile utilizate în sistem scurt, sunt stocate și în sistemul detaliat. Cu toate acestea, într-un sistem detaliat dau descriere detaliata alcătuirea benzilor în cromozomi rearanjați folosind simboluri suplimentare. Coloana (:) indică un punct de pauză, iar două puncte (::) indică o pauză urmată de o reuniune. Săgeata (->) indică direcția de transfer al fragmentelor de cromozomi. Capetele brațelor cromozomilor sunt desemnate prin simbolul ter (terminal), pter sau qter indicând capătul brațului scurt sau, respectiv, lung. Simbolul sep este folosit pentru a indica centromerul.
Tipuri de rearanjamente cromozomiale
Material suplimentar de origine necunoscută. Simbolul add (din latină additio - adiție) este folosit pentru a indica material suplimentar de origine necunoscută care a fost adăugat într-o regiune sau bandă cromozomială. Materialul suplimentar atașat la regiunea terminală va determina o creștere a lungimii brațului cromozomului. La descrierea cromozomilor cu material suplimentar de origine necunoscuta la ambele brate simbolul der este plasat inaintea numarului cromozomilor. Dacă material suplimentar necunoscut este introdus într-un braț de cromozom, simbolurile ins și (?) sunt folosite pentru descriere.
Ștergeri. Simbolul del este folosit pentru a indica ștergerile terminale și interstițiale:
46,XX,del(5)(q13)
46,XX,del (5) (pter->q13:)
Semnul (:) înseamnă că ruptura a avut loc în banda 5q13, ca urmare, cromozomul 5 este format dintr-un braț scurt și o parte din brațul lung, situat între centromer și segmentul 5q13.
46,XX,del(5)(q13q33)
46,XX,del(5)(pter->q13::q33->qter)
Semnul (::) înseamnă o ruptură și reunirea benzilor 5ql3 și 5q33 ale brațului lung al cromozomului 5. Segmentul de cromozom dintre aceste benzi este șters.
Cromozomii derivați sau derivați (der) sunt cromozomi care apar ca urmare a rearanjamentelor care afectează doi sau mai mulți cromozomi, precum și ca urmare a rearanjamentelor multiple într-un singur cromozom. Numărul cromozomului derivat corespunde numărului cromozomului intact, care are același centromer ca și cromozomul derivat:
46,XY,der(9)del(9)(p12)del(9)(q31)
46,XY,der(9) (:р12->q31:)
Cromozomul 9 derivat este rezultatul a două deleții terminale care apar în brațele scurte și lungi, cu puncte de întrerupere la benzile 9p12 și, respectiv, 9q31.
46,XX,der (5)add(5)(p15.1)del(5)(q13)
46,XX,der(5)(?::p15.1-»q13:)
Cromozomul 5 derivat cu material suplimentar de origine necunoscută atașat la banda 5p15.1 și o deleție terminală a brațului lung distal de banda 5q13.
Cromozomi dicentrici. Simbolul mor este folosit pentru a descrie cromozomii dicentrici. Un cromozom dicentric înlocuiește unul sau doi cromozomi normali. Astfel, nu este nevoie să indicați lipsa cromozomilor normali.
45,XX,dic(13;13)(q14;q32)
45,XX,dic(13;13)(13pter->13ql4::13q32-»13pter)
Ruperea și reunirea au avut loc în benzile 13ql4 și 13q32 pe doi cromozomi omologi 13, rezultând un cromozom dicentric.
Duplicări. Dublările sunt indicate prin simbolul dup; pot fi directe sau inversate.
46,XX,dup(1)(q22q25)
46,XX,dup(1)(pter->q25::q22->qter)
Dublarea directă a segmentului dintre benzile lq22 și lq25.
46,XY,dup(1)(q25q22)
46,XY,dup(1) (pter->q25::q25->q22::q25->qter) sau (pter->q22::q25-»q22::q22->qter)
Dublarea inversată a segmentului dintre benzile lq22 și lq25. Trebuie remarcat faptul că doar un sistem detaliat face posibilă descrierea dublării inversate.
Inversiunile. Simbolul inv este folosit pentru a descrie inversiunile para- și pericentrice.
46,XX,inv(3)(q21q26.2)
46,XX,inv(3)(pter->q21::q26.2->q21::q26.2->qter)
Inversie paracentrică, în care ruperea și reuniunea au avut loc în benzile 3q21 și 3q26.2 ale brațului lung al cromozomului 3.
46,XY,inv(3)(p13q21)
46,XY,inv(3)(pter-»pl3::q21->p13::q21->qter)
Inversie pericentrică, în care ruptura și reuniunea au avut loc între banda de braț scurt 3p13 și banda de braț lung 3q21 a cromozomului 3. Regiunea dintre aceste benzi, inclusiv centromerul, este inversată cu 180°.
Inserții. Simbolul ins este folosit pentru a indica inserarea directă sau inversată. O inserție este considerată directă atunci când capătul proximal al regiunii de inserție este într-o poziție proximală față de cel de-al doilea capăt al acestuia. Cu o inserție inversată, capătul proximal al regiunii de inserție este într-o poziție distală. Tipul de inserare (directă sau inversată) poate fi indicat și prin simbolurile dir și, respectiv, inv.
46,XX,in(2)(pl3q21q31)
46,XX,ins(2)(pter->p13::q31->q21::pl3-»q21::q31-qter)
O inserție directă, adică dir ins(2) (p13q21q31), a avut loc între segmentele 2q21 și 2q31 ale brațului lung și segmentul 2p13 al brațului scurt al cromozomului 2. Regiunea cromozomului brațului lung dintre segmentele 2q21 și 2q31 este inserată în bratul scurt in regiunea segmentului 2p13. În noua poziție, segmentul 2q21 rămâne mai aproape de centromer decât segmentul 2q31.
46,XY,ins(2) (pl3q31q21)
46,XY,ins(2)(pterH>pl3::q21->q31::pl3->q21::q31-»qter)
În acest caz, secțiunea inserată este inversată, adică inv ins(2)(p13q31q21). În insert, segmentul 2q21 este mai departe de centromer decât segmentul 2q31. Astfel, locația segmentelor față de centromer s-a schimbat.
Izocromozomi. Simbolul i este folosit pentru a descrie izocromozomii, care sunt cromozomi formați din două brațe identice. Punctele de întrerupere în izocromozomi sunt localizate în regiunile centromerice p10 și q10.
46,XX,i(17)(q10)
46,XX,i(17)(qter-»q10::q10 ->qter)
Izocromozomul de-a lungul brațului lung al cromozomului 17 și punctul de întrerupere sunt desemnate în regiunea 17q10. Cariotipul conține un cromozom normal și un cromozom 17 rearanjat.
46,X,i(X)(q10)
46,X,i(X) (qter-»q10::q10->qter)
Un cromozom X normal și un izocromozom X de-a lungul brațului lung.
Locurile fragile (siturile fragile) pot apărea ca polimorfisme normale sau pot fi asociate cu boli ereditare sau anomalii fenotipice.
46,X,fra(X)(q27.3)
O regiune fragilă din subbanda Xq27.3 a unuia dintre cromozomii X din cariotipul feminin.
46,Y,fra(X)(q27.3)
O regiune fragilă în subbanda Xq27.3 a cromozomului X în cariotipul masculin.
Un cromozom marker (etichetă) este un cromozom modificat structural, din care nicio parte nu poate fi identificată. Dacă se identifică orice parte a unui cromozom anormal, aceasta este descrisă ca un cromozom derivat (der). Când descrieți un cariotip, un semn (+) este plasat înaintea simbolului mar.
47,XX,+mar
Un cromozom marker suplimentar.
48,X,t(X;18)(p11.2;q11.2)+2mar
Doi cromozomi marker în plus față de translocarea t(X;18).
Cromozomii inel sunt desemnați prin simbolul r și pot consta din unul sau mai mulți cromozomi.
46,XX,r(7)(p22q36)
46,XX,r(7) (::р22->q36::)
Ruperea și reunificarea au avut loc în segmentele 7p22 și 7q36, cu pierderea regiunilor cromozomiale distale de aceste puncte de întrerupere.
Dacă centromerul unui cromozom inel este necunoscut, dar segmentele de cromozom conținute în inel sunt cunoscute, cromozomii inel sunt definiți ca derivați (der).
46,XX,der(1)r(1;3)(p36.1q23;q21q27)
46,XX,der(1)(::lp36.1->1q23::3q21->3q27::)
Translocări. Translocații reciproce
Pentru a descrie translocațiile (t), se folosesc aceleași principii și reguli ca și pentru a descrie alte rearanjamente cromozomiale. Pentru a distinge cromozomii omologi, unul dintre omologi poate fi subliniat cu un singur caracter de subliniere (_).
46,XY,t(2;5)(q21;q31)
46,XY,t(2;5)(2pter2q21::5q31->5qter;5pter 5q31::2q21->2qter)
Pauza și reunirea au avut loc în segmentele 2q21 și 5q31. Cromozomii au schimbat regiuni distale de aceste segmente. Cromozomul cu numărul de serie mai mic este indicat mai întâi.
46,X,t(X;13)(q27;ql2)
46,X,t(X;13)(Xpter->Xq27::13ql2->13qter;13pter->3q 12::Xq27->Xqter)
Pauza și reunirea au avut loc în segmentele Xq27 și 13q12. Segmentele distale de aceste zone au fost schimbate. Deoarece cromozomul sexual este implicat în translocare, acesta este înregistrat mai întâi. Rețineți că notația corectă este 46,X,t(X;13), nu 46,XX,t(X;13).
46,t(X;Y) (q22;q1, 1,2)
46,t(X;Y)(Xpter->Xq22::Yq11.2->Yqter;Ypter->Yq11.2::Xq22->Xqter)
Translocarea reciprocă între cromozomii X și Y cu punctele de întrerupere Xq22 și Yq11.2.
Translocațiile care implică brațe întregi ale cromozomilor pot fi înregistrate indicând punctele de întrerupere în regiunile centromerice ale p10 și q10. În translocațiile echilibrate, punctul de întrerupere în cromozomul sexual sau în cromozomul cu un număr de serie mai mic este desemnat p10.
46,XY,t(4;3)(p10;q10)
46,XY,t(1;3)(lpteMlpl0::3ql0->3qter;3pter->3p40::4q40->4qter)
Translocarea reciprocă a brațelor întregi ale cromozomului, în care brațele scurte ale cromozomului 1 s-au unit cu centromerul cu brațele lungi ale cromozomului 3, iar brațele lungi ale cromozomului 1 s-au unit cu brațele scurte ale cromozomului 3.
În translocațiile dezechilibrate ale brațelor cromozomilor întregi, cromozomul rearanjat este desemnat ca un derivat (der) și înlocuiește doi cromozomi normali.
45,XX,der(1;3) (p10;q10)
45,XX,der(1;3)(1pter->1p10::3q10->3qter)
Un cromozom derivat format din brațul scurt al cromozomului 1 și brațul lung al cromozomului 3. Cromozomii 1 și 3 lipsă nu sunt etichetați deoarece sunt înlocuiți cu cromozomul derivat. Cariotipul conține astfel un cromozom normal 1, un cromozom normal 3 și cromozomul derivat der(l;3).
translocații robertsoniene
Acesta este un tip special de translocare care apare ca urmare a fuziunii centrice a brațelor lungi ale cromozomilor acrocentrici 13-15 și 21-22 cu pierderea simultană a brațelor scurte ale acestor cromozomi. Principiile pentru descrierea translocațiilor dezechilibrate care implică brațe întregi se aplică și descrierii translocațiilor robertsoniene folosind simbolul (der). Simbolul rob poate fi folosit și pentru a descrie aceste translocații, dar nu ar trebui să fie folosit pentru a descrie anomalii dobândite. Punctele de întrerupere ale cromozomilor implicați în translocare sunt indicate în regiunile q10.
45,XX,der(13;21) (q10;q10)
45,XX,rob(13;21) (q10;q10)
Ruperea și reunirea au avut loc în segmentele 13q10 și 21q10 ale regiunilor centromerice ale cromozomilor 13 și 21. Cromozomul derivat a înlocuit un cromozom 13 și un cromozom 21. Nu este nevoie să se indice cromozomii lipsă. Cariotipul conține un cromozom normal 13, un cromozom normal 21 și der (13;21). Dezechilibrul apare din cauza pierderii brațelor scurte ale cromozomilor 13 și 21.
Se disting următoarele tipuri de mutații:
A) mutații genomice, ducând la modificarea numărului de cromozomi. Mutațiile genomice apar adesea la plante. În acest caz, poate exista o multiplicare de seturi întregi de cromozomi (poliploidie) sau o creștere (trisomie) sau o scădere (monozomie) a numărului de cromozomi individuali;
b) mutatii cromozomiale(vezi Secțiunea 2.2), în care structura cromozomilor este perturbată, dar numărul lor în celulă rămâne neschimbat. Mutațiile cromozomiale pot fi detectate prin examinare microscopică.
V) mutații genetice, nu conduc la modificări ale cromozomilor care pot fi detectate cu ajutorul unui microscop; aceste mutații pot fi detectate numai prin analiza genetică a modificărilor fenotipice (vezi pct. 3.6).
Studiul mutațiilor la om la nivel de proteine și ADN (în special mutații ale genelor hemoglobinei) a adus o mare contribuție la înțelegerea naturii lor moleculare. Rezultatele acestor studii și rezultatele analizei structurii cromozomiale folosind metode de colorare diferențială de înaltă rezoluție au condus la o estompare a liniei dintre mutațiile cromozomiale și cele ale genelor. Știm acum că ștergerile și inserțiile sunt posibile la nivel molecular și că încrucișarea inegală poate schimba microstructura. Metodele de colorare diferențială au făcut posibilă detectarea la microscop a unor rearanjamente cromozomiale nediferențiate anterior. Trebuie amintit că modificările cromozomiale detectate prin colorare diferențială diferă cu mai multe ordine de mărime
5 Mutații 143
din modificări precum deleţiile genelor structurale. Prin urmare, distincția dintre aberațiile cromozomiale structurale și mutațiile genetice este utilă în scopuri practice.
Celulele în care apar mutații. Cu exceptia tip daune genetice, este extrem de important localizare. Mutațiile pot apărea atât în celulele germinale, cât și în celulele somatice. Cele care apar în celulele germinale sunt transmise indivizilor din generația următoare și, de regulă, se găsesc în toate celulele descendenților care devin purtătorii lor. Mutațiile somatice pot fi detectate numai în descendența celulei mutante corespunzătoare, ceea ce duce la un individ „mozaic”. Consecințele fenotipice vor apărea numai dacă aceste mutații interferează cu implementarea funcțiilor specifice inerente acestor celule mutante.
Frecvențele de mutație. Unul dintre parametrii utilizați cel mai des atunci când se studiază procesul de mutație este frecvență aparitie mutatii(sau rata de mutație). În relație cu oamenii, este definită ca probabilitatea ca un eveniment de mutație să aibă loc în timpul vieții unei generații. De regulă, aceasta se referă la frecvența mutațiilor în ouăle fertilizate. Problema ratelor de mutație în celulele somatice este discutată în secțiune. 5 1.6.





